CRISPR cures a metabolic disorder in an infant's liver
Will personalized gene-editing therapies become the standard of care for ultra-rare diseases?
We’ve heard a lot about CRISPR over the past two decades.
Its use as a genetic engineering tool even won the Nobel Prize in 2020!
But despite all of the promises (and the hype) the use of CRISPR to bring about genome edited cures for patients has been slow.
This is due to a number of factors which include regulatory hurdles, technological limitations, and the uncooperative biology of many rare diseases.
We’re finally starting to get past the regulatory and technological limitations, though, and this is underscored by the recent approvals of CRISPR based therapies for sickle cell disease and β-thalassemia (CASGEVY).
In the early days of CRISPR, which is an acronym for clustered regularly interspaced short palindromic repeats, it was used to create gene knockouts.
What we collectively know as “CRISPR” is actually a combination of a guide sequence and a CRISPR associated protein (usually 9, Cas9) that can recognize and bind to specific pieces of DNA.
Its natural function is to act like a rudimentary immune system in bacteria that recognizes and chops up DNA from phages (bacteria viruses).
But we’ve now been able to co-opt Cas9 and “CRISPR” based guide RNAs to perform genetic engineering!
Over the years, we’ve perfected Cas9 (and other Cas proteins) to be more specific and we can now use them to create personalized therapies that edit the genomes of individuals who inherited misbehaving proteins.
This is much easier to do when those broken proteins function primarily in blood cells or specific organs like the liver because they’re easier to target than trying to fix broken proteins in all of a patients’ muscle cells.
This biological limitation is one that we probably won’t be able to get around (unless we focus on editing out rare diseases from embryos!)
But a personalized CRISPR therapy was recently used to treat an infant with a very rare metabolic disorder.
The infant presented to a clinic with Carbamoyl-phosphate synthetase 1 (CPS1) deficiency.
CSP1 is a metabolic disorder of the liver that results in the buildup of ammonia in the blood and eventually leads to hyperammonemia, seizures and brain damage.
Researchers developed a CRISPR base editing therapy that fixed the targeted mutation (Q335X) and restored the function of CSP1 in mouse models of the disease.
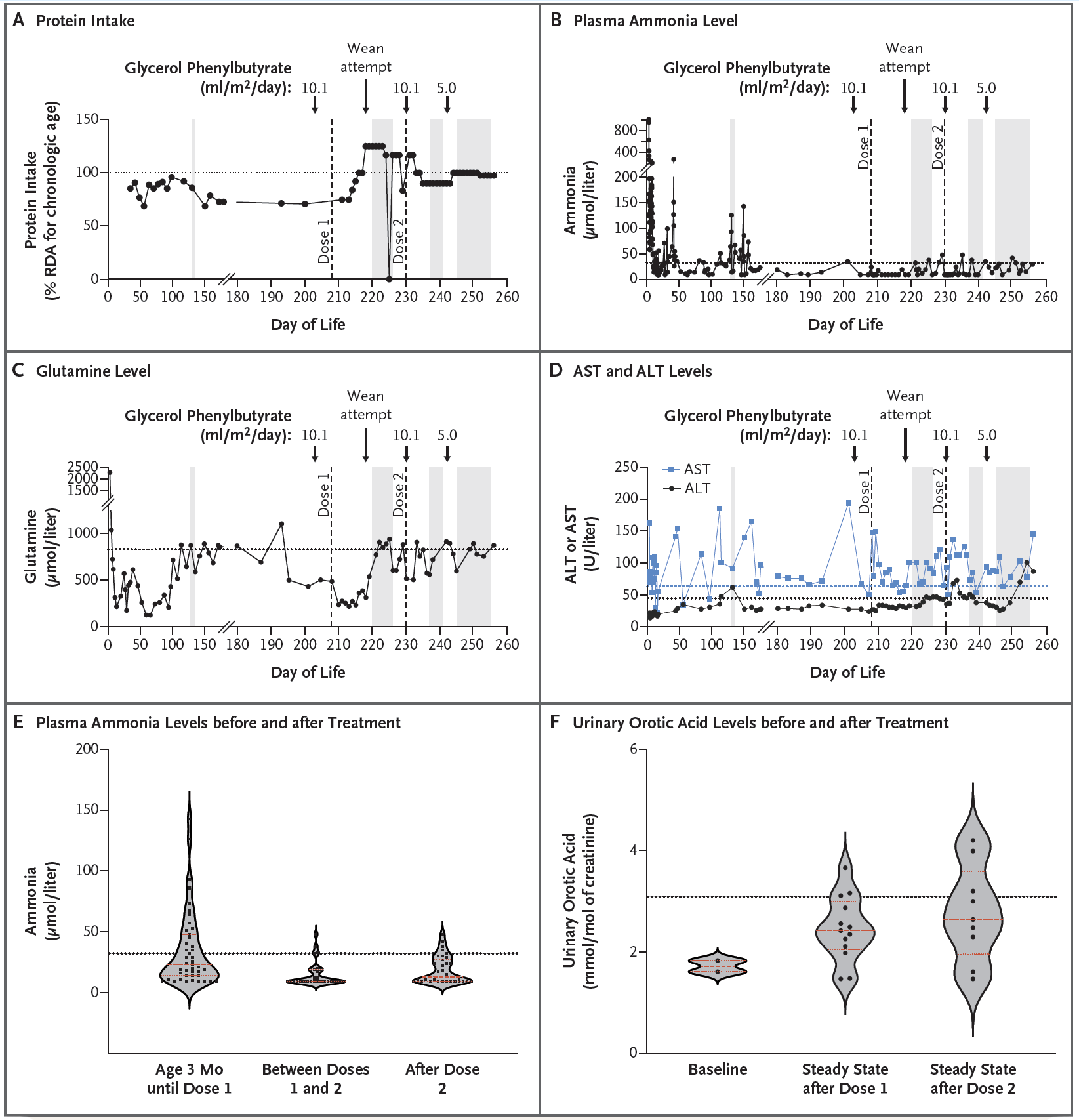
The therapy was delivered to the patient using lipid nanoparticles (which naturally end up being processed by the liver) and made the observations that can be seen in the figure above:
A) Patients with CSP1 deficiency are put on a low protein diet to help mitigate ammonia build up and this chart tracks protein intake over the course of treatment. The patient was weaned off of the low protein diet after the first dose of the therapy
B) After two doses of therapy, the infant’s blood ammonia levels were well stabilized (even under a higher protein diet)
C) AST and ALT levels (liver function) were elevated but not concerning
D) But most importantly, plasma ammonia levels improved significantly after treatment
E) And the patient began excreting more orotic acid which is another indication that the metabolic deficiency was improved by the therapy
The infant who was treated is doing very well (despite suffering a number of unrelated viral infections during the course of the treatment) and requires less supportive care than before the CRISPR therapy was administered.
While more follow-up is required to track the progress of the patient, this is an exciting result that hopefully helps to bring more personalized therapies to the clinic for the hundreds of metabolic disorders that could be similarly treated using CRISPR to fix broken enzymes in the liver!