Omic.ly Weekly 72
April 21, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) Will Ultima's ultra cheap bases be a major boon to MRD?
2) The future of high throughput proteomics is unfolding right in front of us
3) The 'plus and minus' method: Fred Sanger's first attempt at DNA sequencing
Here's what you missed in this week's Premium Edition:
HOT TAKE: Genome interpretation's big week in the spotlight
Ultima Genomics is put to the test in MRD
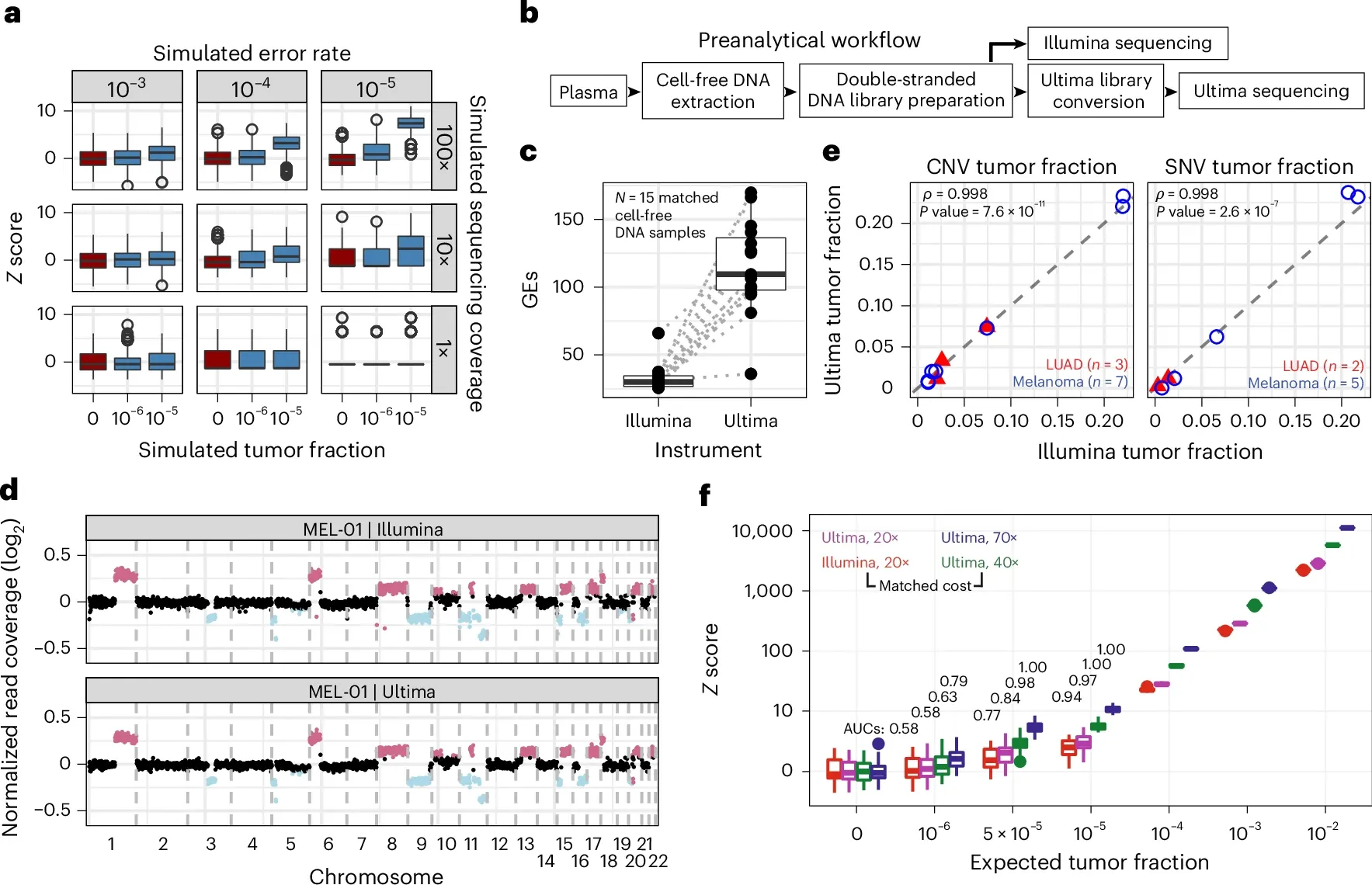
Ultima Genomics came out of stealth in 2022 claiming they were going to upend the sequencing market with a very high throughput instrument that uses silicon wafers to perform the sequencing reaction.
The benefit of Ultima's spinning wafer scheme is that sequencing reagents can be flowed over the entire surface of the wafer and ,unlike Illumina where reagents are discarded every cycle, the Ultima sequencer reuses the reagent mix to decrease costs.
Early reviews of Ultima's UG100 sequencer were mixed because it struggled to sequence homopolymers and the quality was just ever so slightly worse than Illumina.
But they seem to have fixed those problems and now offer a very high throughput system with a cost per base of about half that of its competitors.
This is important because one of the biggest roadblocks to using whole genome sequencing for cancer detection in liquid biopsies has been cost.
A liquid biopsy is just a blood based test that looks for the hallmarks of cancer in the blood.
This is done by sequencing the DNA floating around in the sample to see if we can find fragments with mutations in them.
These mutations can be indicative that someone has cancer, but these fragments are generally very rare, so you have to do A LOT of sequencing to find them!
We've come up with some clever ways to combine tumor informed sequencing (sequence a tumor to find important variants) with targeted capture of tumor mutations to reduce the amount of sequencing we need to do to detect cancer in liquid biopsies.
But it'd be much easier if we could take a blood sample and just sequence it without having to do a bunch of stuff up front!
Since cost is less of an issue on the Ultima platform, researchers at the New York Genome Center thought it might be perfect for performing tumor naive (where you don't sequence the tumor!) minimal residual disease testing (MRD).
MRD is a type of liquid biopsy where you're trying to determine how much cancer is in the body before and after it is treated to monitor when it might recur.
Since detecting recurrence early can have a positive impact on outcome, it's important to detect it as soon as possible which typically means more sequencing is better.
It's a great use case for Ultima's cheap bases!
But before the researchers used the Ultima sequencer for MRD testing, they compared its performance to that of Illumina which can be seen in the figure above. a) is a comparison of sequencing error rates to tumor mutations and shows that to detect tumor fractions below 10-5, you need at least 100x coverage and an error rate of less than 10-4. b) is a workflow diagram showing how they performed the comparison. c) displays a graph of sequencing coverage for each sample. d,e, and f) show that the results from Illumina and Ultima are almost identical with the higher coverage on the Ultima samples (f) showing greater sensitivity for lower frequency variants!
The researchers go on to show that using duplex error corrected reads (ppmSeq) that they're able to use whole genome MRD to track recurrence in a mouse model of melanoma and in patients with urothelial cancer.
This work is clinically important because it's not always possible to get an informative chunk of tumor for performing MRD testing.
In those scenarios, deeply sequencing a patient's blood on a UG100 might be the next best option!
###
Cheng AP, et al. 2025. Error-corrected flow-based sequencing at whole-genome scale and its application to circulating cell-free DNA profiling. Nature Methods. DOI: 10.1038/s41592-025-02648-9
High throughput proteomics could revolutionize our understanding of human disease, but it's going to take some major innovations to get us there!
Proteomics is the study of all of the proteins produced by an organism.
Proteins are:
Enzymes, structural components, signaling molecules, hormones, etc.
They basically do all of the work while DNA and RNA sit around and watch!
But proteins are also the ultimate end product of the information stored in our DNA and are created as a result of two processes:
Transcription - the conversion of DNA into the RNA message.
Translation - the conversion of that RNA message into functional protein.
For decades, DNA and RNA have stolen the show, mostly because innovations in nucleic acid sequencing have made decoding these messages a relatively simple process.
While we can glean some information about the biological status of an organism or tissue by sequencing DNA and RNA, we can actually measure functional biology by looking at the proteins!
And it's the proteins, or lack thereof that cause disease!
So why haven't we heard anything about proteomics?
In some respects, you have.
Many of the diagnostic tests that are administered when you see a physician are measuring protein concentrations or enzyme activity.
Unfortunately, the majority of these only look at a single protein at a time.
But, the real magic lies is in being able to measure all of the proteins!
And not just the select few that we've already found to be clinically relevant!
How can we measure thousands of proteins all at the same time?
We have options:
Mass Spectrometry - There are lots of fancy flavors of mass spec, but in general, this technique works by ionizing proteins, smashing them into a detector, and then imputing what was present before the ionizing and smashing.
Antibody/Aptamer Arrays - An old, low throughput, standby of protein detection has been the use of antibodies. However, innovations like those implemented by Olink which add a sequence tag to antibodies facilitates the multiplex counting of thousands of proteins at a time making this process much more high throughput with a wide dynamic range. A competing technology from Somalogic works similarly, however, they’ve created custom RNA aptamers to do the dirty work.
Protein Sequencing - This is done through a systematic decay and detection process. This involves the labeling of specific amino acids and imputation of the peptide present based on where those labels appear or the use of fluorescent ‘recognizers’ (antibodies) to detect the terminal amino acids throughout the decay process.
These methods can allow us to look at a wide array of proteins, and quantify them (proteomics!), to better understand how changes to the genome affect the biology of our cells and, ultimately, better understand health and disease.
Frederick Sanger invented a famous DNA sequencing method. It's not the one pictured below (which he also invented).
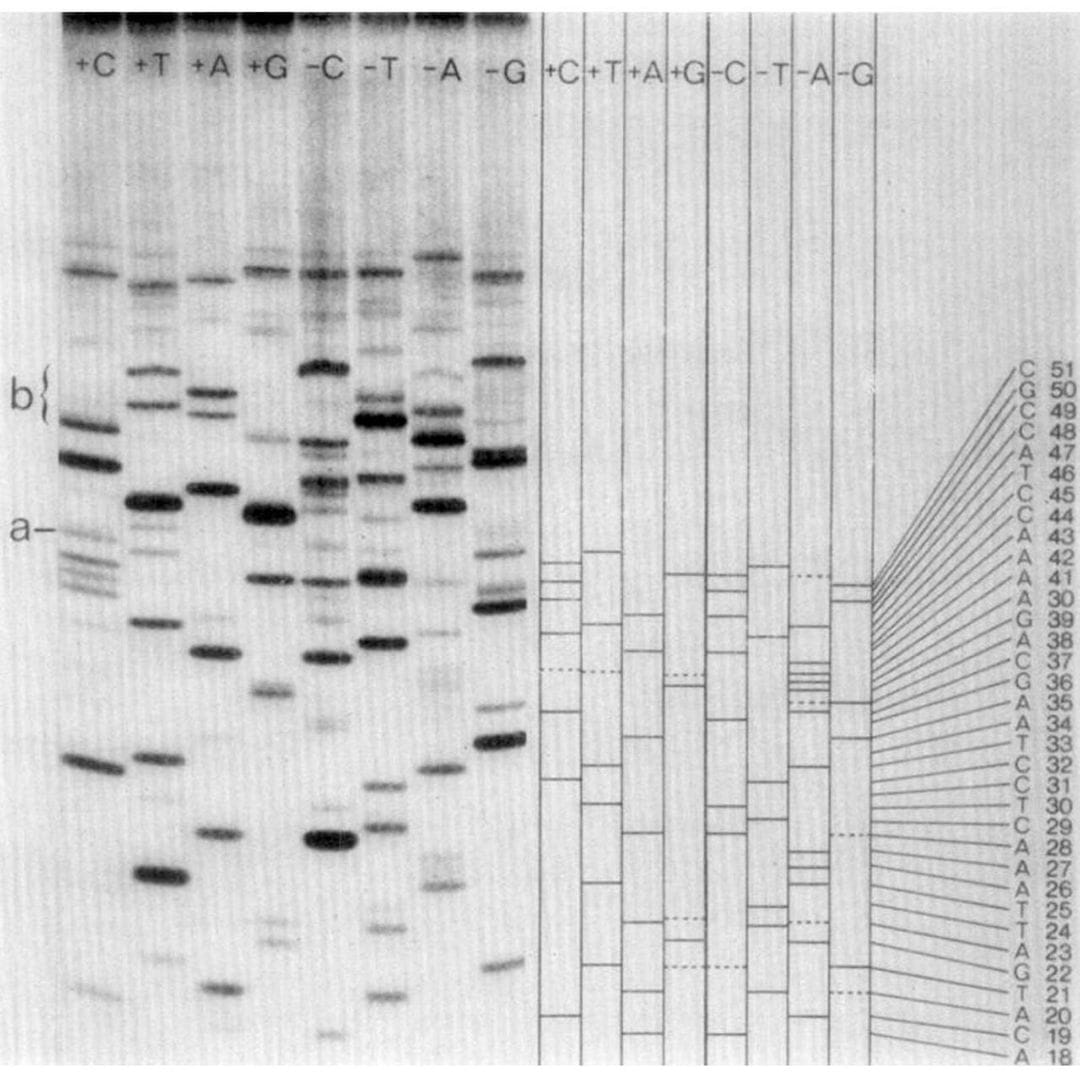
Sanger is one of the few scientists to be awarded more than one Nobel Prize.
His first was for determining the amino acid sequence of Insulin!
His second was shared with Walter Gilbert and Paul Berg for developing nucleic acid sequencing methods.
To this day, 'Sanger' sequencing is a gold standard method in genetics.
And despite the advent of high throughput sequencing technology, millions of Sanger reactions are still performed every year!
But Sanger has another claim to fame.
He sequenced the first DNA genome which was the bacteriophage PhiX174.
This may sound familiar.
Because PhiX is used widely as a sequencing control!
This is partly to memorialize that it was the first, but mostly because it has a similar number of AT and GC bases.
But, Sanger didn't use his eponymous sequencing method to sequence it!
This came as a bit of a shock to me.
And my discovery of this fact went something like this:
“What the (expletive) is the ‘plus and minus’ method!?”
And so, instead of covering Sanger's sequencing of bacteriophage PhiX174.
Instead, we'll review the method he used to do it!
So, what exactly is the 'plus and minus' method?
It can be seen above.
But don't be fooled!
While it looks like regular old gel based Sanger sequencing, there are actually twice as many lanes!
And they're labeled ‘+C+T+A+G’ and ‘-C-T-A-G.’
Are you starting to see where the 'plus and minus' name came from?
The minus lanes are made by priming the thing you want to sequence with a restriction fragment, adding all the bases and a polymerase, and then stopping the reaction.
Those random products are then purified and added to a new reaction that contains only 3 of the 4 bases.
Whatever additional extension occurs stops at the position of the missing base.
So, the band in the gel is -1 from where the -base should be!
The plus lanes are made using a similar process:
Random fragments are generated and then the purified fragments are added to a new reaction containing only 1 of the nucleotides along with T4 polymerase.
T4 polymerase is special because it will 3’ degrade DNA, but interestingly, it will stop degrading DNA at “residues corresponding to the one [nucleotide] that is present.”
Translated: if you only add adenine nucleotides, it creates fragments that all end in adenine.
So, when you run these 8 reactions (+C+T+A+G -C-T-A-G) out on a gel, you can use the plus and minus signals from each base to determine the sequence of the fragment that was present!
Two years later, chain terminating dideoxynucleotides were added to this scheme, allowing 'Sanger' sequencing to be done in 4 lanes instead of 8 and with fewer steps and artifacts.
###
Sanger F, Coulson, AR. 1975. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. JMB. DOI: 10.1016/0022-2836(75)90213-2
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: