Omic.ly Weekly 74
May 12, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) De novo mutations are more common than we previously thought
2) Mass spec is primed to lead the next proteomics revolution
3) The most famous picture in the history of genetics
Here's what you missed in this week's Premium Edition:
HOT TAKE: Elizabeth Holmes' partner looks to raise $50m for a health optimization start-up. What could go wrong?
There’s more to de novo mutations (DNMs) than just SNPs and indels.
Human genetic variation, or the differences in our genomes that contribute to why each of us look, sound, and behave differently, are the result of de novo mutations.
But, before my genetics friends start screaming at me, I just mean that that variation had to be de novo at some point in the past!
Because the textbook definition of a DNM is:
“A mutation or alteration in the genome of an individual organism that was not inherited from its parents.”
DNMs are important because they are the mutations that serve as the basis for the generation of new disease phenotypes, new protein functions and ultimately the evolution of a species.
Because it’s these new mutations that increase or decrease an organism’s ‘fitness’ or ability to survive long enough to produce offspring.
But how do we know if a mutation is de novo or just something that was passed down from one of our parents?
With sequencing!
By looking at how these mutations arise and are transmitted between generations, we can calculate a rate of their occurrence.
We’ve tried to do this before, but because the dominant sequencing technology for the past two decades has been short-reads, we’ve missed important classes of DNMs, which in turn has caused us to underestimate the DNM rate!
These missed mutations include structural variants (SVs), variants in highly repetitive sequences, and variants in things like pseudogenes and paralogs.
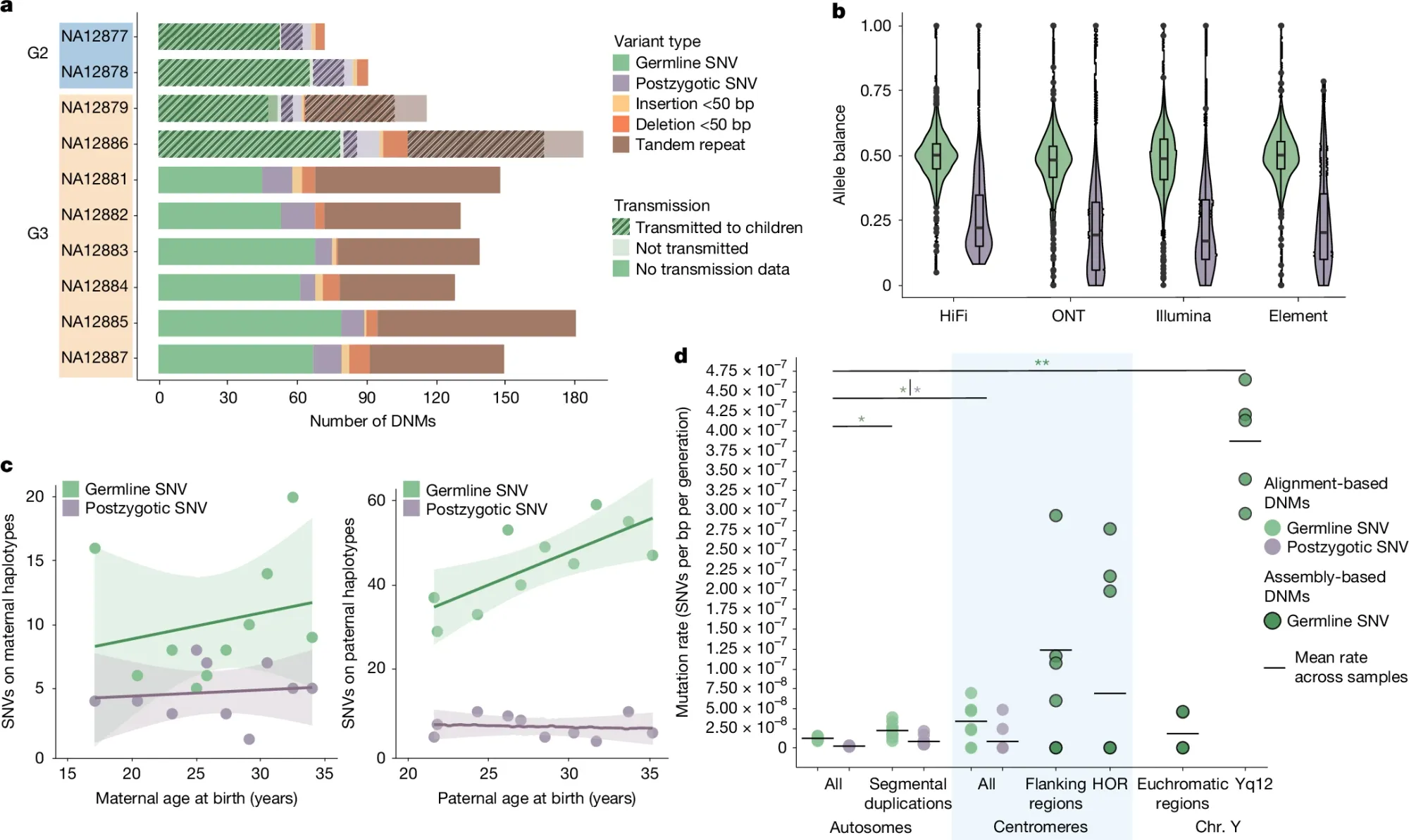
To help get a more accurate rate calculation, researchers have now sequenced the CEPH 1463 family, a well-documented 28-member, four-generation pedigree using both short and long read sequencing technologies to generate near-complete, phased assemblies (>95%) of each family member’s genome.
Part of what they found can be seen in the figure above which focuses on SNP and indel mutations: a) displays the different variant classes found across each generation, b) shows the allele balance of germline variants (green) vs post zygotic mutations (purple - happen after fertilization during development) c) Highlights that these mutations occur more often with parental age and that fathers are responsible for significantly more of them than mothers and d) that there’s significant enrichment of these events in the repetitive regions of chromosomes near the centromeres.
The paper goes on to calculate DNM rates for tandem repeats, SVs, and Y chromosome variants.
The researchers “estimate a range of 98–206 DNMs per transmission (average of 152 per generation) and observe a strong paternal de novo bias (70–80%) and an increase with advancing paternal age, not only for SNVs but also for indels and SVs, including TRs.”
This is an increase from the previously calculated DNM rate of 60-70 per generation and this work underscores how improvements to genome sequencing technology can help us to better understand the complexities of human genetic variation.
###
Porubsky D, et al. 2025. Human de novo mutation rates from a four-generation pedigree reference. Nature. DOI: 10.1038/s41586-025-08922-2
Mass Spec is leading the charge in bringing high-throughput proteomics to a lab near you.

It's one of the most important techniques in proteomics!
Proteomics is the study of proteins, how they function and how they interact with other molecules within our cells and tissues.
Understanding what all of those proteins are doing is important.
But not just for our own education!
Malfunctioning, missing, or improperly regulated proteins are generally what cause disease.
And quantifying what proteins are present or how they are regulated with post-translational modifications (PTMs) can tell us a lot about how they're functioning in a cell or tissue!
But, there are a number of ways we can analyze proteins.
These can include using antibodies, sequencing them directly, or detecting them with mass spectrometry (mass spec).
While the last one might sound intimidating, it’s currently the most informative of the 3!
And recent innovations have allowed these instruments to detect thousands of proteins in a single sample, including their PTMs which are important on/off switches for proteins!
Knowing that a protein is present is useful, but knowing how it is modified tells you a whole lot more about what it's doing.
Mass spec is currently the only technique capable of reliably getting both of these forms of data in a single output.
And it’s able to do this by ionizing proteins and smashing them into a detector to determine their mass-to-charge ratio or m/z.
This m/z information can then be used to figure out the original chemical composition of the things that were smashed!
There are a couple different flavors here that are worth mentioning:
LC-MS(/MS) - Liquid chromatography - tandem mass spectrometry (two detectors) is probably the simplest form of mass spec used in proteomics. LC here is used to crudely separate proteins before they’re ionized and smashed.
MALDI-TOF (Matrix assisted laser desorption/ionization - time-of-flight) - A sample is combined with a ‘matrix’ that allows it to be more completely ionized. Time-of-flight (a long tube) is added to this scheme to better separate ions of different masses (the big ones take longer to hit the detector) and gives superior resolution over traditional LC-MS! Electrospray Ionization (ESI) is a related but gentler ionization method.
timsTOF - Adds another fun twist to time-of-flight mass spectrometry by sticking a gas tube ahead of the TOF tube to trap ions and selectively release them based on their m/z to increase resolution even further.
zenoTOF - Similar to above but is an ion trap that pulses ions into the TOF tube.
Orbitrap - This is something totally different although the readout is the same and it’s the highest resolution option (also the most expensive)! There’s no smashing in this setup (WTF?) but the system is able to determine the m/z of the ionized proteins as they travel in an elliptical path around a detector.
The most famous photo in the history of genetics wasn’t generated by Watson and Crick, but that didn’t stop them from using it to solve the structure of DNA.
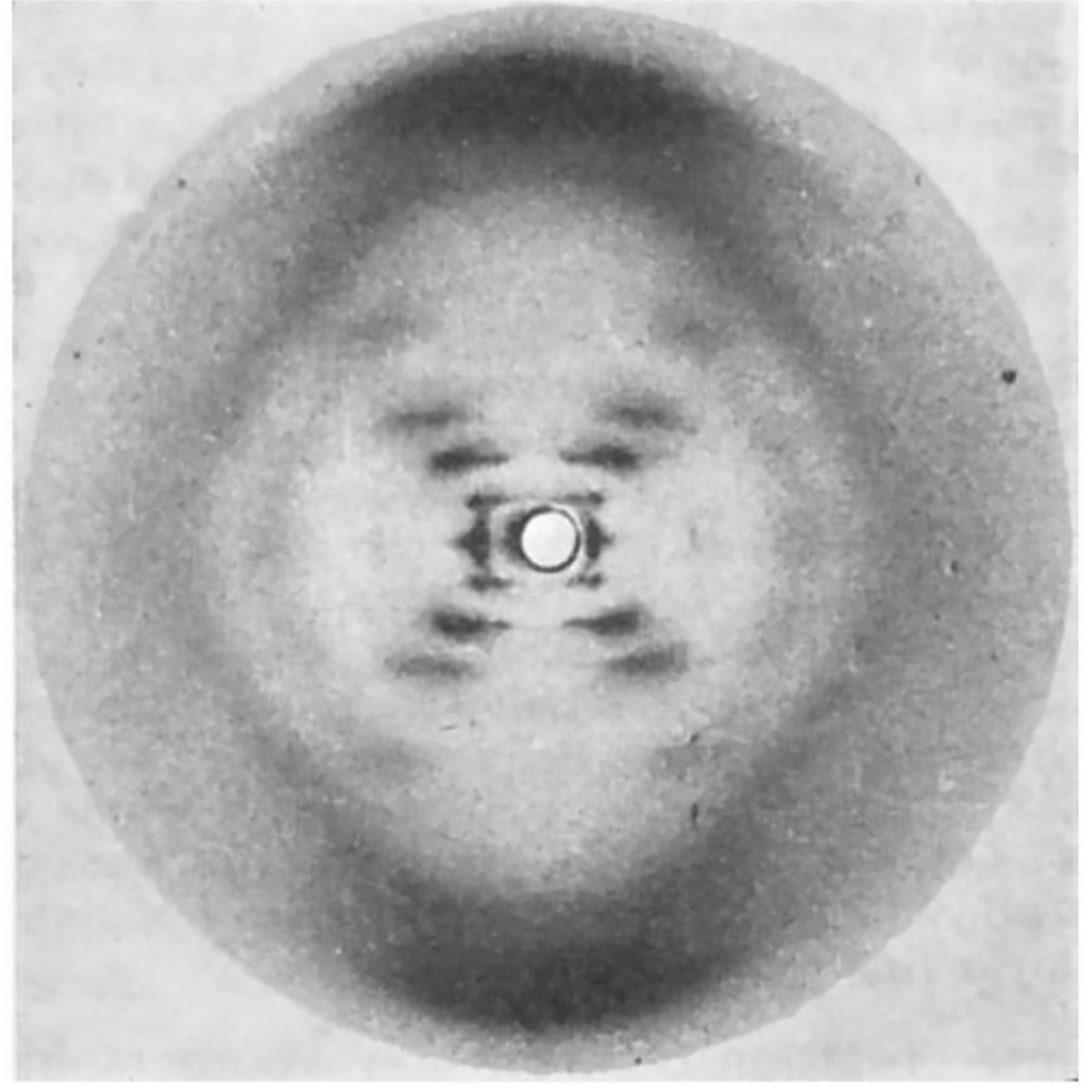
While they tend to get all of the credit for this structure, 3 papers were published back-to-back on this topic in April of 1953:
1) Watson and Crick’s has a single, hand-drawn, 3D structure of DNA.
2) Wilkins' includes a blurry diffraction pattern.
3) Franklin and Gosling's, which can be seen above, has the pristine diffraction pattern of DNA known as Photo 51. Their analysis showed that the structure is helical, it's double stranded, and the bases face inward with the phosphate backbone on the outside.
So, why are there 3 papers?
In the early 1950’s, scientists were coming around to the idea that DNA was the genetic material, but the publication of the Hershey-Chase experiment in 1952 removed all doubt.
Soon after, Linus Pauling, an American structural biologist, published a triple-helical structure of DNA (with the bases facing outward).
Unfortunately for Pauling, his structure was based on flawed data but this put the teams in the UK on notice.
Watson and Crick were at Cambridge while Wilkins, Franklin and Gosling were at King’s College London.
Wilkins did much of the early work generating x-ray diffraction patterns of DNA, but it was Franklin and her PhD student, Gosling, who perfected the art.
In May of 1952, Gosling generated photo 51 of B-DNA, but Franklin was more interested in studying A-DNA.
So, photo 51 wasn’t revisited until she decided to leave King’s.
Since Gosling was transferring to Wilkins to complete his PhD, photo 51 was shared with him and he in turn shared it with Watson.
Realizing this fixed all of their problems, he and Crick completed the model for their structure of DNA, culminating in the publication of all 3 papers in 1953.
Unfortunately, Franklin died of ovarian cancer in 1958 and Watson, Crick and Wilkins won the Nobel in 1962.
The story of photo 51 is incredible, but what’s even more incredible is that Franklin’s contribution was nearly forgotten.
This changed when Watson’s misogyny got the better of him.
In his 1968 memoir, he composed a very unkind depiction of Franklin (He apologized in a later edition).
This sparked renewed interest in her and corrected portrayals of her and her work have been published:
She was a pioneer of x-ray crystallography.
She provided the key diffraction pattern for the structure of DNA.
She resolved the structure of RNA in Tobacco Mosaic Virus.
And, prior to her death, she worked on polio virus.
Her collaborator, Aaron Klug, finished this project and was awarded the Nobel Prize, in part, for his crystallography work.
Though short a Nobel (or two), Rosalind Franklin's scientific legacy is extraordinary.
###
Franklin RE and Gosling RG. 1953. Molecular Configuration in Sodium Thymonucleate. Nature. DOI: 10.1038/171740a0
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: