Omic.ly Weekly 76
May 26, 2025
Hey There!
If you were previously a paid subscriber, I've made it about 3/4 of the way through giving everyone partial (or full...) refunds.
I should be able to finish that up this week. Stripe and my bank haven't been wanting to cooperate, but we're getting there!
Stay tuned to LinkedIn for the job update (If you're interested!)
As always, thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) CRISPR cures a metabolic disorder in an infant's liver
2) Feeling like the dog that caught a car now that you have all this proteomic data?
3) PCR has a more interesting history than you might have been told
4) Weekly Reading List
Will personalized gene-editing therapies become the standard of care for ultra-rare diseases?
We’ve heard a lot about CRISPR over the past two decades.
Its use as a genetic engineering tool even won the Nobel Prize in 2020!
But despite all of the promises (and the hype) the use of CRISPR to bring about genome edited cures for patients has been slow.
This is due to a number of factors which include regulatory hurdles, technological limitations, and the uncooperative biology of many rare diseases.
We’re finally starting to get past the regulatory and technological limitations, though, and this is underscored by the recent approvals of CRISPR based therapies for sickle cell disease and β-thalassemia (CASGEVY).
In the early days of CRISPR, which is an acronym for clustered regularly interspaced short palindromic repeats, it was used to create gene knockouts.
What we collectively know as “CRISPR” is actually a combination of a guide sequence and a CRISPR associated protein (usually 9, Cas9) that can recognize and bind to specific pieces of DNA.
Its natural function is to act like a rudimentary immune system in bacteria that recognizes and chops up DNA from phages (bacteria viruses).
But we’ve now been able to co-opt Cas9 and “CRISPR” based guide RNAs to perform genetic engineering!
Over the years, we’ve perfected Cas9 (and other Cas proteins) to be more specific and we can now use them to create personalized therapies that edit the genomes of individuals who inherited misbehaving proteins.
This is much easier to do when those broken proteins function primarily in blood cells or specific organs like the liver because they’re easier to target than trying to fix broken proteins in all of a patients’ muscle cells.
This biological limitation is one that we probably won’t be able to get around (unless we focus on editing out rare diseases from embryos!)
But a personalized CRISPR therapy was recently used to treat an infant with a very rare metabolic disorder.
The infant presented to a clinic with Carbamoyl-phosphate synthetase 1 (CPS1) deficiency.
CSP1 is a metabolic disorder of the liver that results in the buildup of ammonia in the blood and eventually leads to hyperammonemia, seizures and brain damage.
Researchers developed a CRISPR base editing therapy that fixed the targeted mutation (Q335X) and restored the function of CSP1 in mouse models of the disease.
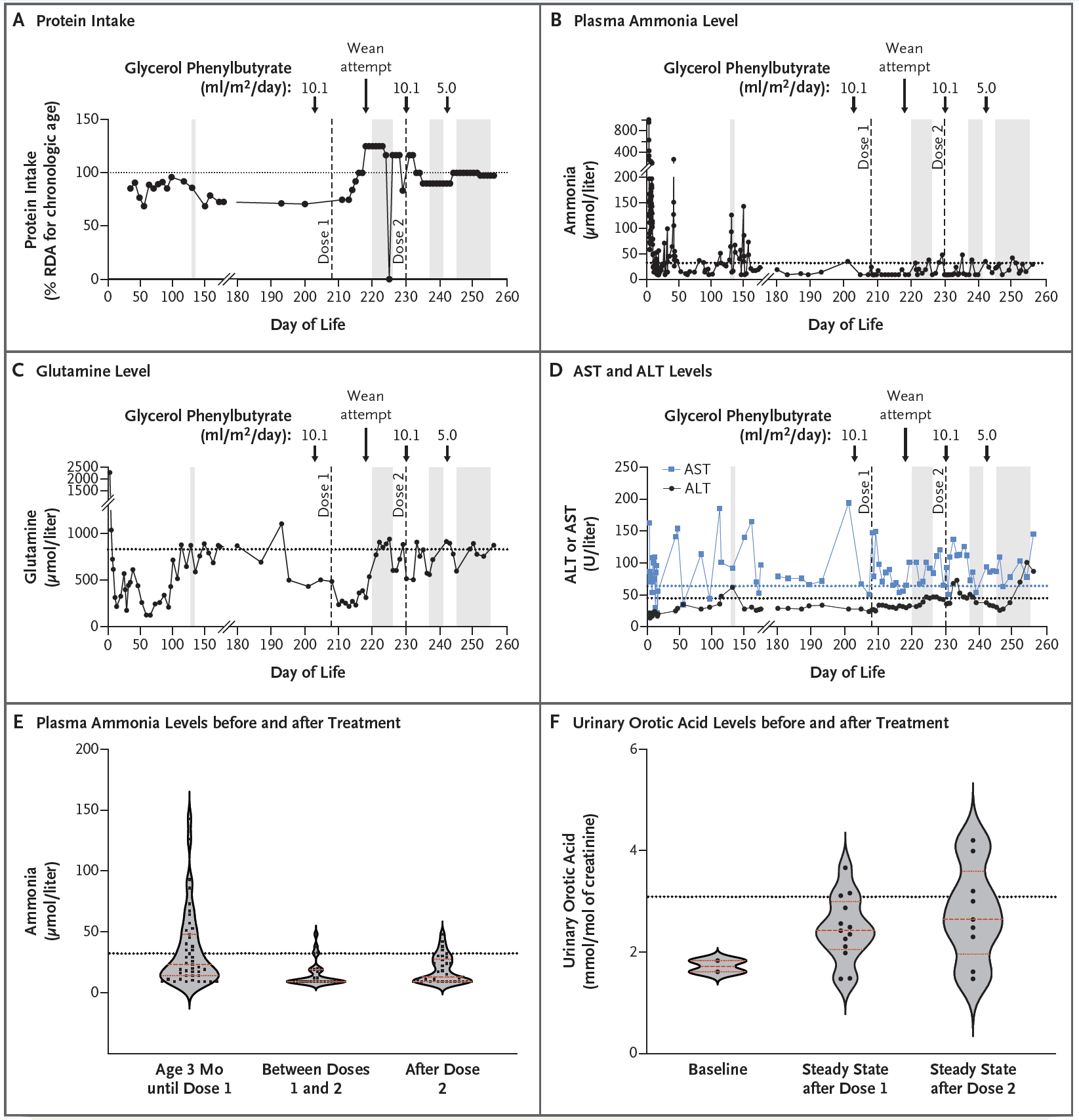
The therapy was delivered to the patient using lipid nanoparticles (which naturally end up being processed by the liver) and made the observations that can be seen in the figure above:
A) Patients with CSP1 deficiency are put on a low protein diet to help mitigate ammonia build up and this chart tracks protein intake over the course of treatment. The patient was weaned off of the low protein diet after the first dose of the therapy
B) After two doses of therapy, the infant’s blood ammonia levels were well stabilized (even under a higher protein diet)
C) AST and ALT levels (liver function) were elevated but not concerning
D) But most importantly, plasma ammonia levels improved significantly after treatment
E) And the patient began excreting more orotic acid which is another indication that the metabolic deficiency was improved by the therapy
The infant who was treated is doing very well (despite suffering a number of unrelated viral infections during the course of the treatment) and requires less supportive care than before the CRISPR therapy was administered.
While more follow-up is required to track the progress of the patient, this is an exciting result that hopefully helps to bring more personalized therapies to the clinic for the hundreds of metabolic disorders that could be similarly treated using CRISPR to fix broken enzymes in the liver!
###
Musunuru K., et al. 2025. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. NEJM. DOI: 10.1056/NEJMoa2504747
So, once we have all of this amazing proteomic data, what do we do with it???

That's a fantastic question!
But your first question might actually be 'What the heck is proteomics!?'
Proteomics is defined as the study of proteins, their functions, regulation and interactions within an organism.
While the genome holds all of our genetic information, the proteome is the genome in action.
And studying the proteome is quite a bit different than studying the genome because the genome is mostly static.
We have no idea what the impact of a mutation or a variant within the genome will be until we see it manifest as a phenotype (a visible symptom, trait or characteristic).
We can see these things on the molecular level by looking at what proteins are produced!
We can figure this out using a variety of techniques including immuno affinity arrays, mass spectrometry and, in the future, protein sequencers.
But once we've gathered the data, what do we do with it and how can we use it to learn anything?
That answer really depends on the experiment that was performed to generate the data. For clinical applications of proteomics I see 3 types of studies being really important:
Longitudinal studies: it's a big word but it just means looking at how things change over time. For example, these could be used to monitor treatment response in oncology patients or detect flares in Crohn's patients.
Case-control studies: these compare diseased individuals to healthy individuals or, diseased tissues to healthy tissues - looking for differences between the two that could be indicative of health or disease.
Single cell studies: look at how proteins or their interactions change from cell to cell to get a more granular and nuanced view of tissue function, treatment response, or disease presentation.
Analysis is focused on looking at changes over time, among disease states or across tissues.
But a key first step in doing any of these analyses is normalization!
You want to be sure you're comparing apples to apples and that the differences you see aren't just because of some bias that was introduced.
There are a couple of options here, a popular one is to use a protein that is commonly expressed at a static level.
Once everything is normalized you can start digging in!
Differential protein expression: compare how protein levels change from dataset to dataset. These are usually visualized as heat maps.
Pathway analysis: determine what proteins are present, how they're modified, and how they interact with one another. These are visualized as networks or more recently as circos plots.
But one of the biggest drawbacks of doing proteomics is that we're still creating a knowledge base.
Our techniques for looking at the proteome historically have been very low throughput.
Thankfully, that's changing, and new initiatives are helping to provide proteomic references we can use to better hone our analyses!
The method below has been cited more than 600,000 times and is one of the most important developments in the history of science.
The polymerase chain reaction (PCR) is used widely to amplify DNA sequences.
Kary Mullis is often given sole credit for developing PCR, which, according to him, he discovered while "riding DNA molecules" during an acid trip in the early 1980's.
But, today's story begins a decade earlier in the lab of Har Gobind Khorana.
Khorana won the Nobel Prize in 1968 for his work figuring out how RNA codes for protein.
The key to his success was that he and his team synthesized their own molecules of RNA, called oligonucleotides.
This allowed them to see what amino acids ended up in proteins after the translation of their sequences.
Khorana continued to focus on scaling this synthesis process to create a full gene sequence.
One of his post-docs, Kjell Kleppe, had an idea to use little pieces of complementary DNA to kickstart these synthesis reactions and copy a target sequence with DNA polymerases.
At the time, it was known that polymerases were involved in copying DNA during cell division and could use a 'primer' to start this reaction.
He presented his method and initial data in 1969 and published a paper where his method of in vitro "DNA repair replication" was described in 1971.
So, why does Mullis get all of the credit for the discovery of PCR?
Mostly because of timing.
In 1970, the Khorana lab was one of the few groups making oligonucleotides.
Kleppe's result was seen as interesting but technically infeasible to scale and the true power of DNA copying wasn't recognized until the early 1980's when cloning and other molecular manipulations were really taking off.
Mullis was employed at that time by Cetus Corporation and he stumbled on the idea of copying DNA using primers and polymerases all on his own.
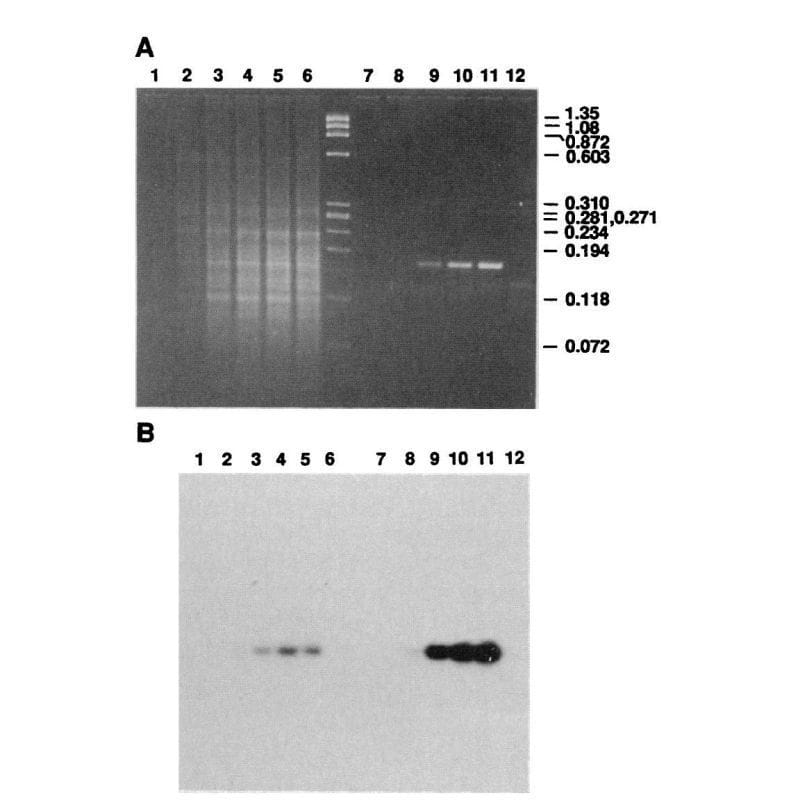
He published an initial paper on PCR in 1985 using the E. coli ‘Klenow’ fragment (a truncated portion of DNA polymerase) which required manually cycling the reaction in a water bath and adding back enzyme every round because it wasn’t heat stable.
However, Mullis realized he could use the polymerase from a different bacteria, T. aquaticus, which lives in the boiling hot springs of Yellowstone Park.
This polymerase, which we now call Taq, was heat stable and didn't require replacement between cycles!
The figure above is Mullis' comparison of Klenow (Lanes 2-5) to Taq (Lanes 8-11).
(A) is an agarose gel and (B) is a southern blot using a radioactive probe to detect the targeted DNA.
The benefits of using Taq are pretty obvious, but its heat stability was game changing because it made the process much cleaner and easy to automate!
Mullis received the Nobel Prize for this work in 1993.
###
Saiki RK, et al. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. DOI: 10.1126/science.2448875
Weekly Reading List











Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: