Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) CRISPR-Associated Transposases (CASTs) make large DNA insertions in Human cells a reality
2) The basics of Proteasomes, Lysosomes, and the therapeutics that can target them!
3) A DNA diffraction pattern that was lost to history
4) Weekly Reading List
Tired of only being able to use CRISPR to make small edits? CASTs are here to change that!
Genome editing technologies have improved significantly since the early days of plasmid preps and restriction digests.
That's especially true now with the emergence of programmable nucleases like CRISPR.
These tools enable precise modification of short DNA sequences (<200 bp) and have shown some clinical utility in treating genetic disorders.
But efficiently inserting large DNA sequences (≥1 kb), or even complete genes, at specific genomic sites in human cells is still a challenge.
This limitation is significant because sometimes base editing or CRISPR inactivation of a gene aren’t enough and to really fix a problem we need to replace an entire gene!
CRISPR, unfortunately, can’t do this but can do something similar by triggering double-stranded DNA breaks (DSBs) to promote gene insertion via homology-directed repair (HDR) or alternative methods like homology-independent targeted integration (HITI).
However, HDR is inefficient in non-dividing cells and HITI lacks control over orientation and copy number.
DSBs can also cause harmful genomic byproducts such as indels, large deletions, and chromosomal translocations which makes using CRISPR for large genomic insertions infeasible.
To overcome these issues, researchers have explored CRISPR-associated transposases (CASTs), a bacterial system that allow the insertion of kilobase-scale DNA fragments using Tn7-like transposons.
Type I-F CASTs are particularly attractive due to their high insertion specificity and efficiency in bacteria.
But transferring this activity to human cells has proven difficult because these proteins have very low activity in human cells!
To address this, the researchers used phage-assisted continuous evolution (PACE) to evolve a CAST system that has better performance in human cells.
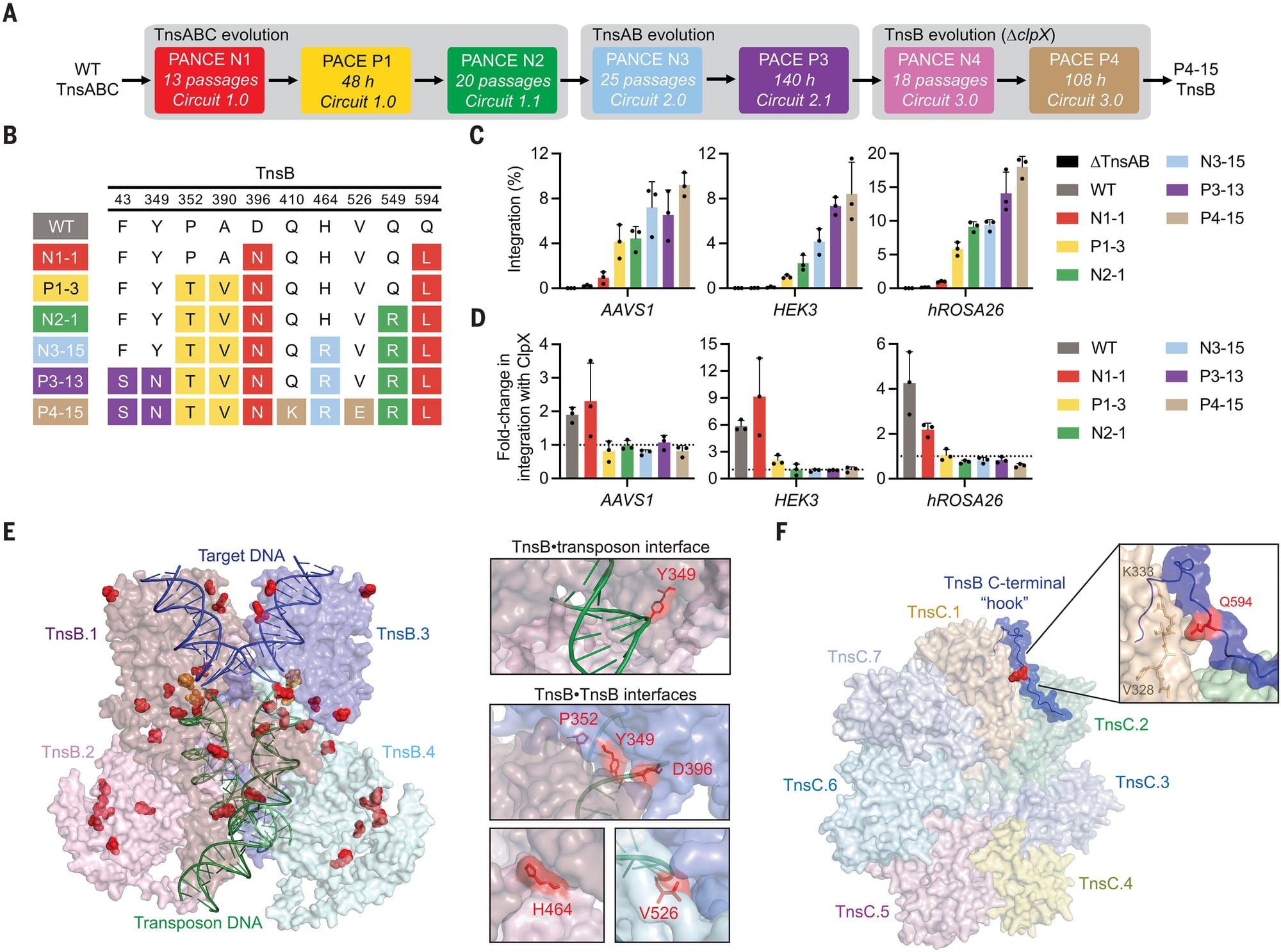
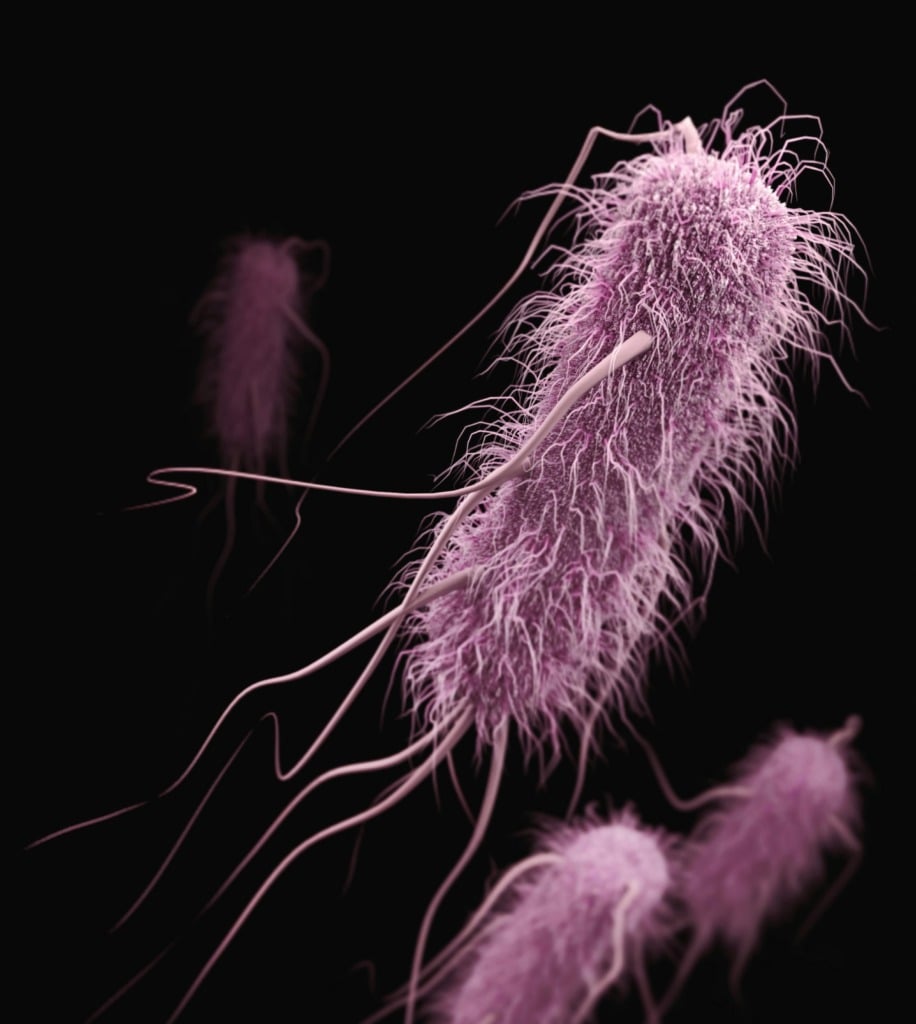
The results of this work can be seen in the figure above:
A) a schematic of the evolution process
B) a table of the mutations found in the best performing transposases
C) shows how well each new tranposase works (higher percent integration is better)
D) the new tranposases don’t require the helper protein ClpX to function efficiently
E and F) space filling models of the engineered proteins highlighting how the mutations interact with DNA.
This evolved system, named evoCAST, achieved up to 30% targeted insertion efficiency at 14 different genomic loci in human cells which is orders of magnitude better than the wild-type system.
CASTs provide an interesting new twist on genome engineering, adding a new tool in our toolkit to help bring more tailored and personalized treatments to the clinic.
###
Witte IP, et al. 2025. Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science. DOI: 10.1126/science.adt5199
PROTACs and LYTACs: How therapeutics can take advantage of proteomic garbage men.
This is especially true when things go awry!
Cells have mechanisms in place to degrade misbehaving proteins, eliminate things that aren’t needed anymore, or respond to crises by going nuclear.
For cells, the nuclear options are autophagocytosis (self-eating), senescence (cell cycle arrest), apoptosis (programmed cell death), and necrosis (rapid, less programmed, cell death).
But, in some diseases these processes are interrupted and unruly proteins are allowed to continue their delinquent behaviors.
Things can get even worse when cells that should have pushed the red button and sacrificed themselves for the good of everyone else, don’t.
So, how can we use proteomics to get these cells back on track (or obliterate them)?
Well, cells have two distinct protein waste removal services that we can take advantage of:
Proteasome - These are like protein wood chippers that float around inside a cell. They’re used to chop up useless, misfolded, damaged, or no longer needed proteins!
Lysosome - Is a specialized membrane enclosed organelle (kind of like an industrial garbage dump) where bulk protein degradation activities take place.
Targeted protein degradation using these two pathways has been an important area of therapeutic development in the last few years.
This work has focused on the creation of ‘chimeric’ molecules that combine the function of two or more molecules together to perform a desired function:
PROteolysis TArgeting Chimeras (PROTACs) - The proteasome degradation pathway is activated when the proteasome comes across proteins that have been tagged with a molecule called ‘ubiquitin.’ This tag is added to proteins by an enzyme, E3 ubiquitin ligase. So, most PROTACs are composed of two components, one that binds the protein we’d like to degrade, and another that binds E3! This brings that protein into very close proximity to the enzyme that marks things for destruction - bada bing, bada boom - no more protein!
LYsosomal TArgeting Chimeras (LYTACs) - The lysosome degrades lots of things including food that’s been brought in from outside the cell (phagosomes), receptors that once resided on the cellular membrane (endosomes), and larger cellular structures like mitochondria (autophagosomes). How all of those things get to the lysosome is pretty well known, and chimeric molecules can be engineered to cause target proteins to be scooped up in one of those ‘-somes’ and degraded in the lysosome! Just like PROTACs, these chimeric combinations involve binding to a target of interest on one end, and to a lysosomal targeting protein/molecule on the other!
PROTACs and LYTACs have shown promise as adept therapeutic garbage men in inflammatory disease (degrade misbehaving receptors), neurodegeneration (target plaque forming proteins), and cancer (initiate cell death).
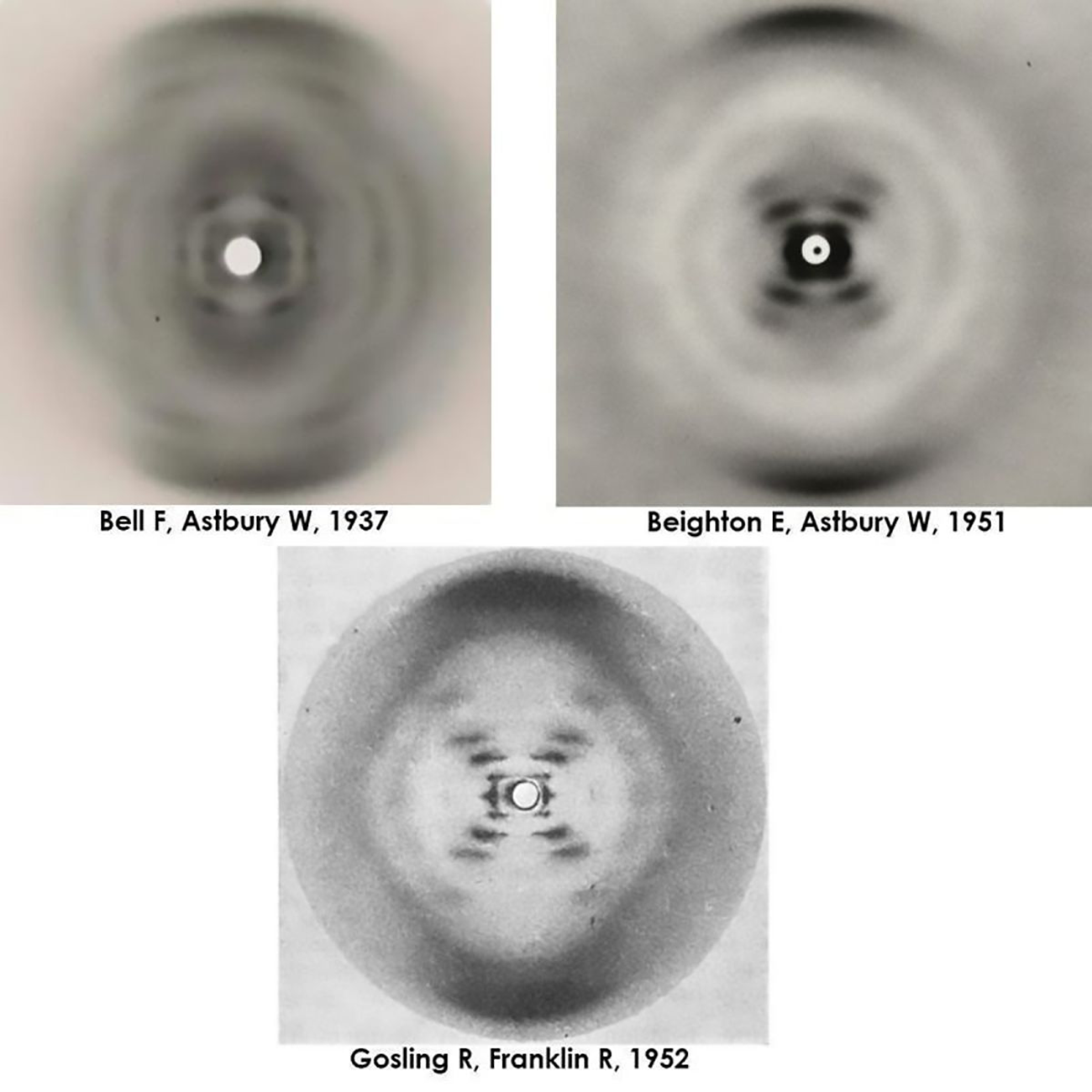
Elwyn Beighton and William Astbury generated a nearly flawless diffraction of B-DNA in 1951, a full year ahead of Franklin and Gosling. They never shared or published it.
As with everything on the path to the discovery of the DNA double-helix, timing was everything!
Well, timing, and knowing what you're looking at.
William Astbury might not be a name you've heard before but he's considered a founder of molecular biology.
He was one of the first to use x-ray diffraction to study protein structures.
He was a protege of William Bragg who in 1915 won the Nobel Prize, along with his son Lawrence, for the discovery that x-rays could be used to determine the location of atoms within a molecule.
What the Bragg's found was that if you blasted crystals with x-rays, the x-rays bounced off of the atoms in those molecules to create specific patterns on x-ray film.
Working backwards, they realized they could deduce from those patterns the structure of the underlying molecule!
However, by 1926, Bragg got bored with blasting simple molecules and tasked his graduate student, Astbury, with studying larger fibrous biological molecules like wool.
Since textiles were an important commodity, studying their properties and how to modify them to improve their commercial value was a big deal.
This led Astbury to start a lab in 1928 in textile physics where he made a name for himself diffracting just about every biological fiber in existence.
So, it should be no surprise that in 1937 he turned his attention to the most important biological fiber of all, DNA.
He, along with a talented graduate student, Florence Bell, created the first ever diffractions for DNA and they published the first proposed structure for DNA, what they referred to as a 'pile of pennies,' in 1938.
Unfortunately, Bell and Astbury's work on DNA was cut short by World War II and Astbury didn't return to the problem of DNA until the late 1940's.
But by 1951, Elwyn Beighton, a lab tech turned graduate student, had picked up where Bell left off and produced a nearly perfect diffraction of DNA.
Unbeknownst to anyone at the time, DNA could take on two forms: a totally dehydrated A form, a hydrated B form, or a mix of both depending on the humidity during drying.
The figure above actually shows 3 figures.
It has Bell's original diffraction of DNA (likely a mix of A and B), Beighton's significantly improved diffraction, and finally, photo 51, Franklin and Gosling's pristine diffraction of B-DNA.
All of the 'what ifs' aside, it's thought that Astbury was too preoccupied with protein structures to recognize the importance of Beighton's cruciform DNA diffraction.
The image was never published, and was lost to history.
Fortunately, the world only had to wait 2 additional years for the structure of DNA to be solved in 1953.