Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) Fuzzy Sequencing: Sometimes close enough is good enough
2) The "Microbiome" isn't just one thing
3) How Linus Pauling used electrophoresis to characterize sickle cell anemia in 1949
4) Weekly Reading List
DNA is coded by 4 bases, but no one said that's how we have to sequence it. Ready to have your mind bent?
My brain was never more broken than when I tried to learn about SOLiD's color space but "Fuzzy Sequencing" is a close contender!
If you remember the ABI SOLiD sequencer, it operated VERY differently than the sequencing by synthesis technology that has come to dominate sequencing for the past 2 decades.
SOLiD was a "sequencing by ligation" technology that ligated fluorescently labeled nucleotide pairs together to determine the sequence of a target DNA molecule.
It also employed an incomprehensible color encoding scheme where 4 dyes were used to label the 16 (4x4) nucleotide pairs and the sequence was determined by deconvoluting the sequences obtained using 3 different sequencing primers (each shifted by one base).
If that sounds confusing, it is - but it helped introduce this concept that just because there's 4 bases, it doesn't mean you have to sequence them individually in a strictly linear fashion.
Oxford Nanopore (ONT) has done something similar with its sequence detection scheme where their pores aren't actually sequencing individual bases at any given time...they're predicting what Kmer (an ~ 6 base sequence) is present in the pore.
It's much easier to try to detect a Kmer than to detect the signal of a single base as it occludes a nanopore!
But both SOLiD and ONT use(d) their sequencing schemes to try to identify an accurate sequence of the individual bases contained in the DNA fragment being sequenced.
What if you didn't care what the sequence actually was?
Are there more efficient ways to identify a target molecule than by sequencing it at single base resolution?
Those might sound like insane questions, but some of the most lucrative applications of sequencing technology DO NOT require single base resolution to give us the answers we're seeking.
For example, most pathogen detection or microbial identification tests, non-invasive prenatal testing for chromosomal anomalies, or whole transcriptome profiling - basically anything where we're counting/detecting fragments - don't need single base resolution to accurately identify those things!
So, it could make sense to create a new sequencing technology that encodes genetic information more cost effectively and efficiently for those kinds of applications.
Enter Fuzzy Sequencing!
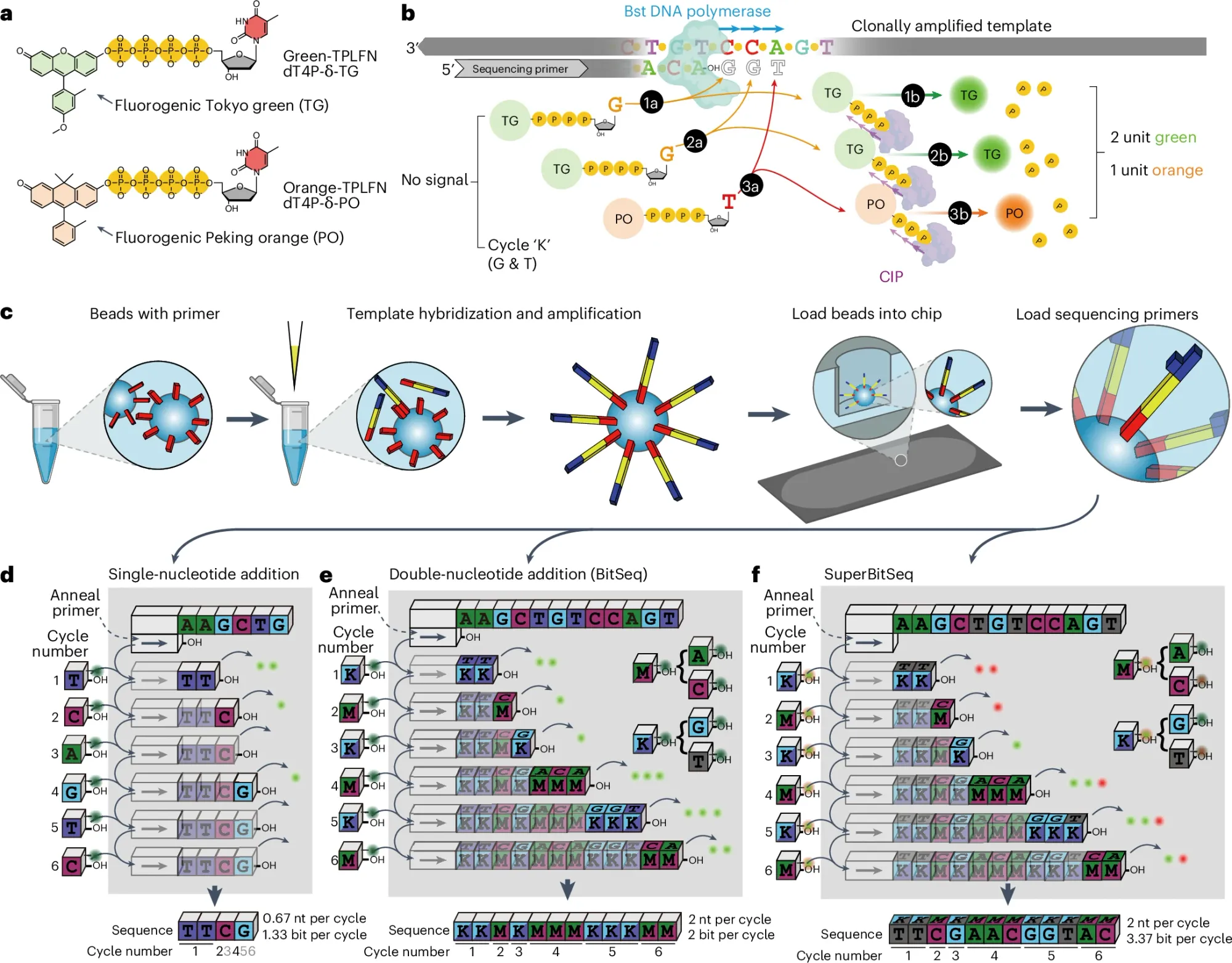
Instead of reading out individual bases, Fuzzy Sequencing provides a "Fuzzier" read out which is depicted in the figure above not as bases but as an encoded flowgram! (see d)
a) It uses one (BitSeq) or two (SuperBitSeq) fluorgenic dyes
b) tagged bases are added to each cycle as 'flowgrams' (eg K = G/T and M = A/C) are flowed together and unlike Illumina sequencing that uses reversible terminators, this chemistry does not, so multiple bases can be added every cycle (similar to how IonProton pyrosequencing works) but that's ok because the number of bases added can be determined by the fluorescence intensity of the signal.
c) the sequencing reaction is based on bead emulsion PCR where beads capture a single sequence, amplify it on the bead surface and then that bead is deposited in a nanowell for the sequencing reaction - here the sequencing cycles proceed by sealing and unsealing the wells with oil to prevent reagent diffusion or signal mixing
d) compares the sequencing efficiency of a single base addition per cycle to that of BitSeq and SuperBitSeq - which is to say that adding multiple bases per cycle is faster and cheaper with a readout of not A,C,T, and G but as K and M!
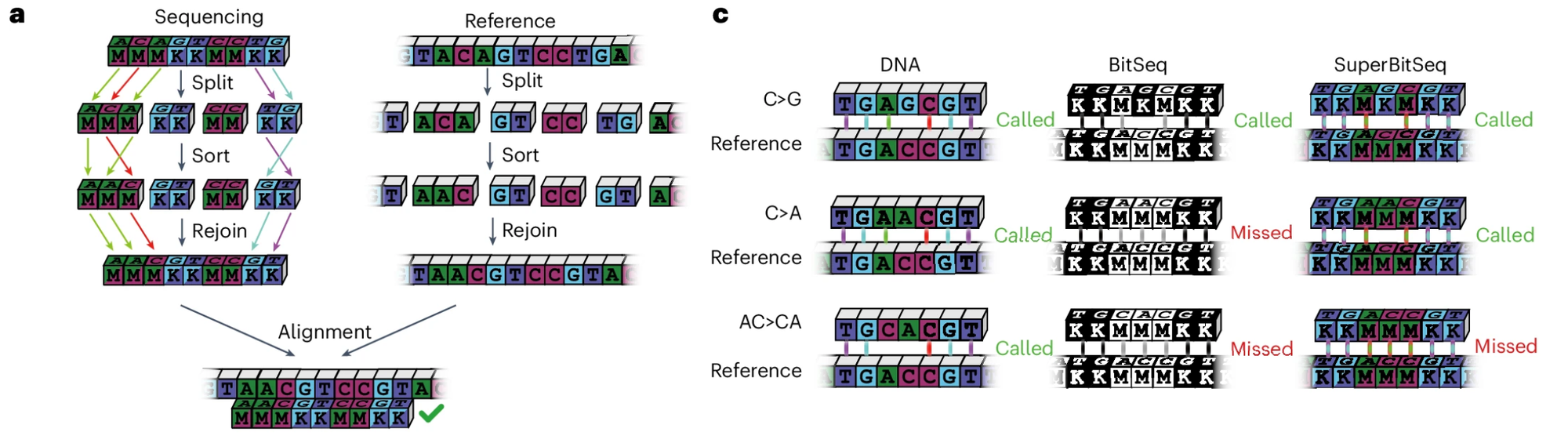
And if you're having trouble understanding how this encoding works to identify a genetic sequence, the researchers provided this fun diagram:
Obviously encoding information this way means that there will be base ambiguity at some positions (see 'missed' above), but for the applications where you'd be using this, you don't care!
The researchers show in follow-up experiments that Fuzzy Sequencing is able to accurately detect large copy number variations in prenatal testing cases (including chromosomal microdeletions) and identify microbes from throat and anal swabs.
While I don't see Fuzzy Sequencing taking the sequencing community by storm, it's an interesting thought experiment and if developed commercially could be a nice alternative to multiplex digital or real-time PCR.
It could also make prenatal testing, transcriptomics, and even oncology testing significantly more cost effective!
“The microbiome” isn't just one thing. It's lots of things and each habitat has its own.
To say the human microbiome is complicated is an understatement.
It's generally agreed that a microbiome is the community of organisms (bacteria, fungi, viruses, archaea) that live and interact in a particular habitat inclusive of the environmental, physical and chemical properties of that habitat.
This means that we're surrounded by microbiomes and if we're talking about them from the human perspective, there are at least 6 that we need to discuss:
Respiratory tract - The upper airway, your nose and the cavity behind it are colonized by specific microbes that thrive in the mucosa that helps to warm, humidify and filter the air that we breathe. Similarly, the lower respiratory tract and lungs host their own crew of microbes. The microbiomes in these regions change significantly during respiratory infections or after the development of respiratory diseases like asthma.
Eye - Anyone who has ever suffered from conjunctivitis (infection of the eye) understands the importance of having a healthy eye microbiome. But the eye has three distinct habitats which include the outer eye, the conjunctiva (the membrane that covers the eye), and the meibum (the fatty, slippery, liquid that lubricates our eyes!). Each of these are inhabited by specific microbes that thrive in these ocular environments!
Oral - Our mouth is one of the microbiomes we’re most conscious of. Dental hygiene is extremely important for human health, and a dysfunctional oral microbiome is typified by unpleasant odors as a result of infections of our gums and teeth.
Skin - This is one of the more complicated microbiomes to classify since “skin” is present on every outer surface of our body. The skin microbiome can differ significantly depending on whether we’re talking about our armpit, scalp, or face! But be aware, this surface that you scrub clean every day is host to trillions of microbes!
Urogenital - This primarily refers to the vaginal microbiome which changes throughout a woman’s cycle and during pregnancy. The microbial community here is sensitive to changes in pH (and can cause changes in pH!). Dysfunction can lead to infertility or even miscarriages.
Digestive - The “gut” microbiome is probably the one that we’ve heard the most about. But the digestive tract is made up of the stomach, small intestine, large intestine, and colon. Each of these has a distinct physical and chemical environment, and you guessed it, each supports its own unique microbial community!
While we contain multitudes of microbes that form communities on and in various parts of our body, we’re still at the earliest stages of trying to tease out the cause and effect relationships here that impact human health.
While it’s pretty easy to define an unhealthy microbiome when we have an infection, identifying the factors that create and maintain a healthy one is still a huge work in progress!
Sickle Cell Anemia was the first inherited disease to be molecularly characterized. It was done in 1949 using a revolutionary new method: electrophoresis.
Sickle cell anemia affects 4.4m people, and 43m are carriers of the trait.
It is characterized by the crescent, or sickle shape, of the red blood cells of those affected by the disease.
James Herrick first discovered sickle-shaped blood cells in a patient suffering from severe anemia in 1910.
Through subsequent observation it was realized that there was an asymptomatic form of the disease, sickle cell trait.
In those individuals it appeared that they had a mixture of normal and sickle blood cells.
Further study within the families of these individuals in 1923 revealed that sickle cell was hereditary or passed down from parents to their offspring.
And because those with sickle cell trait appeared to have a 50/50 mix of sickle/normal blood cells, it was determined that this was a recessive Mendelian disease.
Linus Pauling, a titan of early molecular biology, was no stranger to blood or the protein hemoglobin and spent many years in the 1930’s studying hemoglobin’s interactions with oxygen.
Pauling had a suspicion that the structure of proteins played a vital role in their function and was first introduced to sickle cell anemia in 1945.
He hypothesized that the sickling of cells could be related to a change in the structure of hemoglobin since red blood cells are literally just bags that contain a boat load of hemoglobin protein.
So he and his team, Harvey Itano and John Singer, tried to figure out a way that they could show that a difference in the structure of hemoglobin was the cause of sickle cell anemia.
After a bit of trial and error, they stumbled on the use of electrophoresis, a brand-new technique at the time, that allowed for the separation of molecules based on their electrical charge.
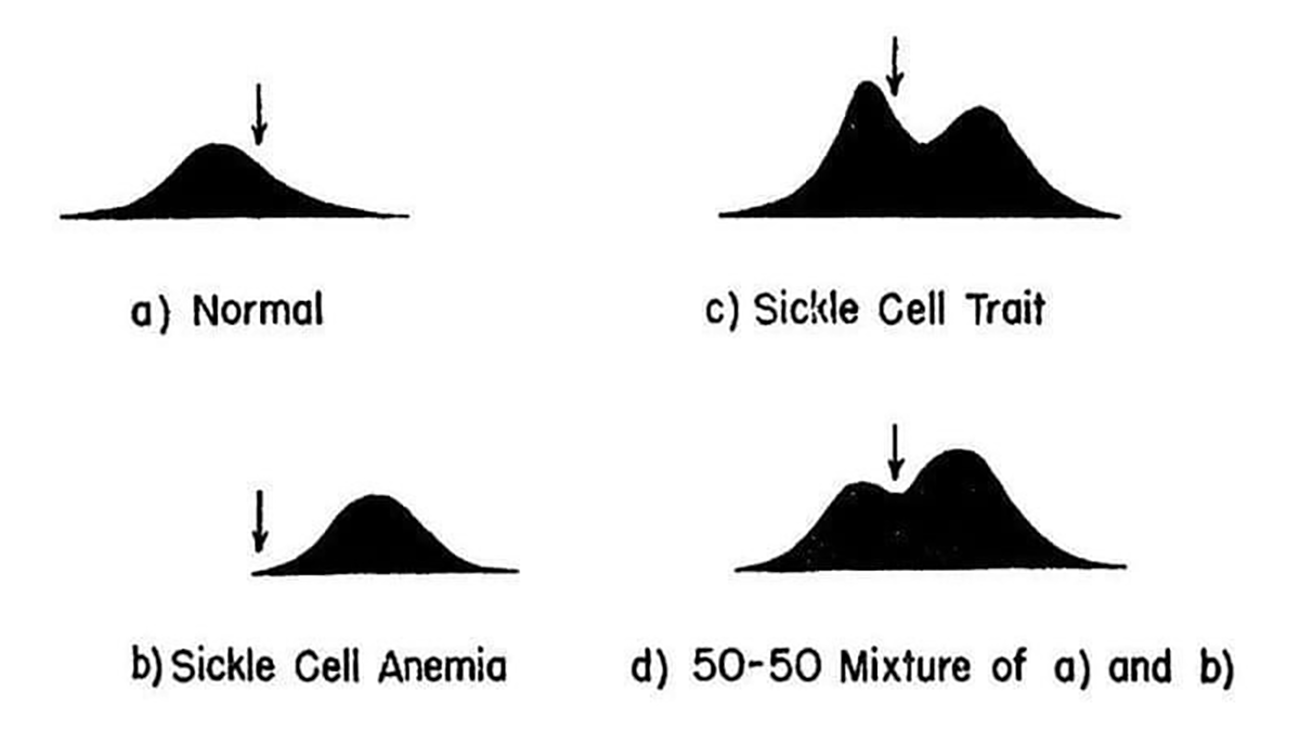
The results of their experiments can be seen in the figure below.
They separated and quantified 4 sets of blood samples using Longsworth scanning diagrams. A) shows normal hemoglobin, B) is hemoglobin from a sickle cell patient, C) is hemoglobin from a patient with sickle cell trait, and D) is a mixture of A and B. The arrow denotes a point of reference for comparing the diagrams.
This work demonstrates that there is a molecular basis for sickle cell anemia and that changes to a gene can alter the structure of a protein.
In the case of sickle cell, this functional relationship extends further because in the 1950’s, the trait was shown to be protective of malaria.
This explains evolutionarily why this disease is found in individuals of African descent; however, it fueled an unfounded fear of 'black blood' throughout the early 1900's.
###
Pauling L et al. 1949. Sickle Cell Anemia, a Molecular Disease. Science. DOI: 10.1126/science.110.2865.543