Omic.ly Weekly 13
February 25th, 2024
Hey There!
Thanks for spending part of your Sunday with Omic.ly!
This week's headlines include:
1) The human genome sequence is finally (almost) totally complete. The pesky Y chromosome was the last hold-out!
2) Multiplex aptamer/antibody arrays are a gateway drug to get genetics people to embrace proteomics
3) Fred Sanger, the father of DNA sequencing, cut his teeth doing protein sequencing and snagged a Nobel in the process!
Please enjoy!
What you missed in this week's Premium Edition:
HOT-TAKE: Complete Genomics wants America to give them a second chance. Should they?
The human Y-chromosome has finally been fully sequenced! No, seriously this time, the genome is actually finished now, maybe.
You might be asking yourself why we keep hearing about the completion of the human genome.
It's like the scientific gift that keeps giving and we're probably going to continue reading these stories because nothing in science is ever really finished.
The more we dig, the more we find, and that presents new questions and generates new hypotheses!
So why'd it take so long to finally sequence the Y chromosome?
Well, over half of it is composed of highly repetitive sequence that couldn't be resolved with short-read sequencing or Sanger sequencing.
Gaps have persisted due to the complex architecture of Y, including palindromes and repetitive sequences which make assembly of these sequences challenging.
Fortunately, in the decades since the genome was 'finished' the first time, we've developed new long-read methods that can span these complicated regions and they were used in combination with short-read polishing to generate the latest reference.
Short of a couple small regions that still need some attention, it's finally done!
This is important because the human Y chromosome and its SRY locus is what determines sex in mammals (whether you are biologically male or female).
This is of particular interest in the case of evolution and sex determination because a number of non-mammalian species have lost their Y chromosome and this latest work could help us to understand how mammals could suffer a similar fate.
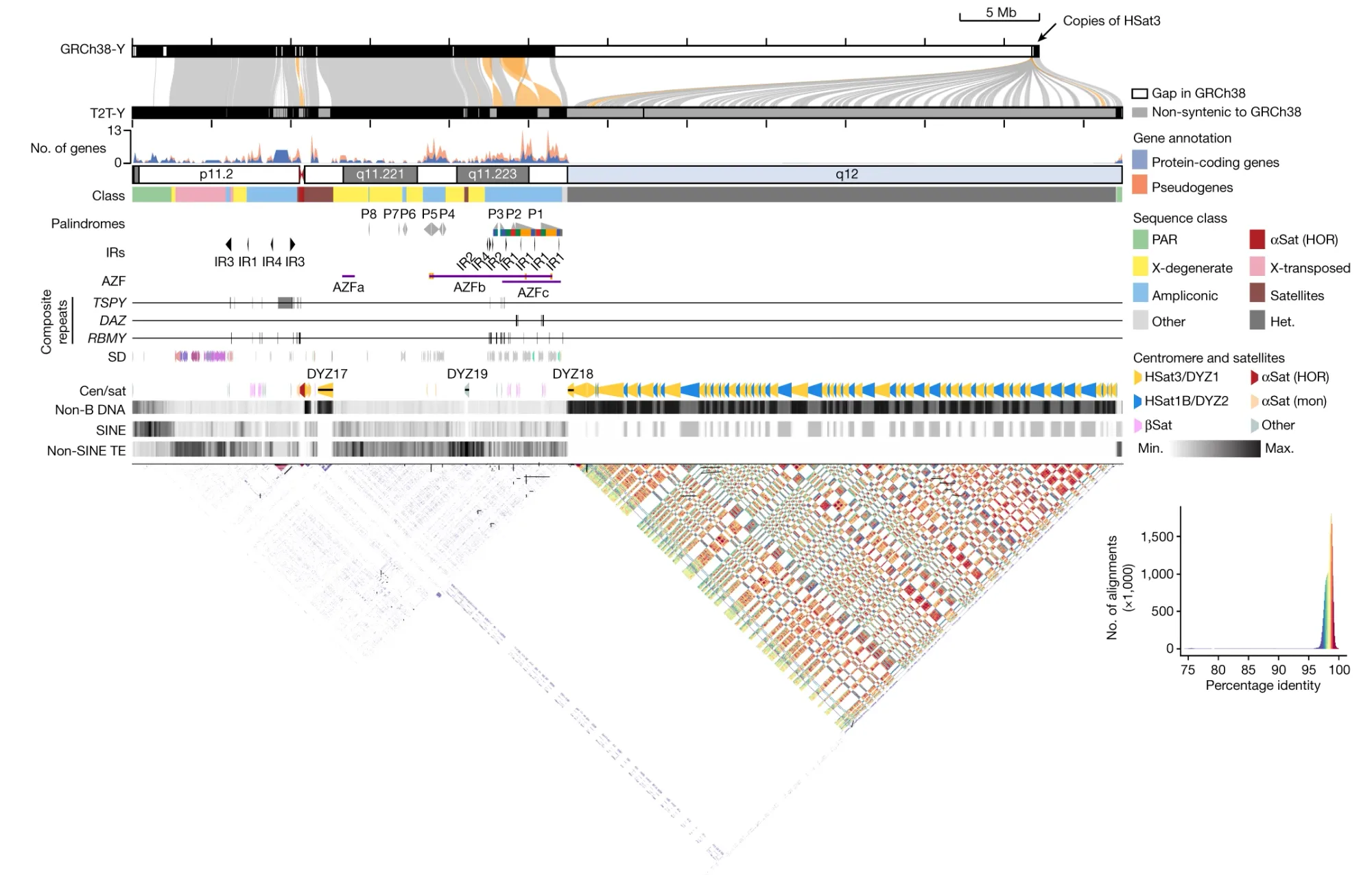
To that end, the Telomere-to-Telomere (T2T) consortium successfully assembled the complete Y chromosome of HG002, T2T-Y.
The final assembly was 62,460,029 bases in length and shows approximately 30 Mb of previously uncharacterized sequence, predominantly from the heterochromatic region of the q-arm.
The researchers further analyzed the gene content, repeat sequences, centromeric regions, and other features of T2T-Y.
They identified, "an additional 110 genes, among which 41 are predicted to be protein coding. The majority of these protein-coding genes (38 of 41) are additional copies of TSPY."
The paper went on to confirm that the number of copies of TSPY and a handful of other repetitive genes can vary greatly between individuals (a second paper in Nature dives into this in-depth).
The researchers also characterized new complex structural features on the Y chromosome (see the figure below), and improved variant calling for XY individuals using T2T-Y as a reference, showing enhanced accuracy in variant detection and reducing the number of false-positive variant calls.
The completion of T2T-Y and the availability of complete genome references from diverse populations are the cornerstones of advancing our understanding of human genetics.
###
Rhie A, et al. 2023. The complete sequence of a human Y chromosome. Nature. DOI: 10.1038/s41586-023-06457-y
Multiplex aptamer/antibody arrays are a gateway drug to get genetics people to embrace proteomics.

When we completed the human genome in 2003, it was heralded as the dawning of a new age in disease prevention.
The truth is that finishing the genome was the first answer in a much longer FAQ about all of the things we didn’t understand in biology.
And since we’ve mostly sorted out the basic code of our genomes, we now need to answer the questions about what all of those genes are doing!
We can do this by looking at other -omes, like the proteome.
But one of the trickiest parts here is that the translation of our genetic code into protein is a massive amplification of that original genetic signal!
There are two copies of each of your genes.
When those are converted into an RNA message, it’s usually not just one RNA that’s made, it can be hundreds.
And then those hundreds of messages can make many thousands of proteins.
So, in genetics we’re usually differentiating the 2 alleles of a gene, but in proteomics we can sometimes be looking for a handful of proteins in a sea of 10,000,000,000!
What this means is that we have to be able to look at way more molecules in proteomics to get the answers we’re looking for than we’ve ever had to look at in genomics.
And it also means the dynamic range is important, or, can the method we’re using detect very abundant and very rare things.
In proteomics we need 10 orders of magnitude, or 1:10,000,000,000 to be the most comprehensive.
We can get pretty close to that using techniques like mass spectrometry, but mass spec instruments are expensive and complicated and most genetics people are infatuated with their sequencers.
So, how can we scam them into doing proteomics while still making them feel like they’re getting good use out of their sequencers?
We can make them use those instruments as counting devices!
Which is a perfect match for two techniques that allow us to detect 10,000+ proteins at a time with the appropriate amount of dynamic range.
Enter Multiplex Aptamer and Immuno Affinity Arrays:
SomaLogic - They do the detecting with aptamers, which are just single stranded pieces of DNA that have been selected for their ability to bind to specific proteins. Detection of said proteins is done by sequencing and counting the aptamers that ended up getting stuck to the proteins in a sample.
Olink - Are more traditional and use antibodies. But these aren't any old antibodies! They've been tagged with DNA! And it's those DNA tags that are then detected and counted on a sequencer.
Both techniques are great at detecting and counting proteins.
But, the major drawback here is that they aren’t able to easily detect protein modifications which are key indicators of the functional state of an expressed protein.
Because of this, they’re probably just a bridge, but for now, they’re perfect for getting people excited about the power of proteins!
Fred Sanger received a Nobel Prize for his work with Insulin. As the father of DNA sequencing, this surely was for insulin's nucleic acid sequence? It wasn't.
DNA sequencing was his second Nobel.
His first was for the amino acid sequence of insulin.
To say Sanger was an accomplished scientist is an understatement and, unfortunately, his early work sequencing insulin doesn't get nearly the attention that it deserves.
But the experiments leading up to this Nobel worthy achievement began in 1943.
As a new member in Charles Chibnall's group at Cambridge, it was suggested that Sanger focus on exploring the amino acid composition of insulin.
At the time, insulin was basically the only highly purified protein available due to its use in treating diabetes.
It was pure luck that insulin was a small protein, but, as is usually true, small packages can be deceiving, and it took 12 years for him to determine its complete sequence.
The majority of this work was done by tagging proteins on the N-terminus using fluorodinitrobenzene (FDNB) followed by acid hydrolysis and/or trypsin digestion and then separation in two dimensions - first by electrophoresis and then by paper chromatography.
Or, more simply, FDNB turns proteins yellow. Sanger then chopped them into smaller fragments, and the separation technique allowed him to count which amino acids appeared in each fragment and to deduce their order.
The final protein sequence was stitched together by lining up all the overlapping sequence fragments.
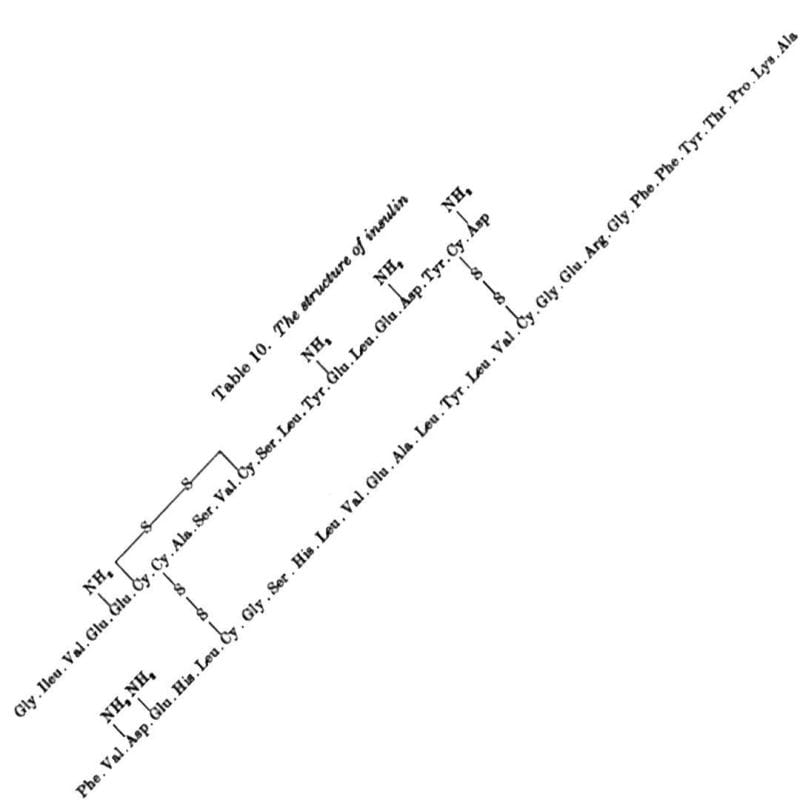
Sanger discovered Insulin has an A chain and a B chain. He sequenced the B chain first, and published that 30 amino acid sequence in 1951.
The 21 amino acid sequence of the A chain wasn't completed until 1952, mostly because it has an intramolecular disulphide bridge that made sequencing it difficult.
The figure above is the culmination of years of hard work that had to be completely redone using slightly different methods that played nicer with disulphide bridges, but this allowed for the identification of their location within the sequence.
The figure below shows the amino acid sequence of both A and B chains of insulin along with the 3 disulphide bridges (S-S) - one intramolecular within A and two others that connect chain A to chain B.
Importantly, this work settled a debate about the structural nature of proteins which, among some scientific circles, was believed to be somewhat fluid.
Sanger showed that insulin had a specific amino acid sequence and, by extension, this was likely true for all proteins.
This seemingly minor detail is what set the stage for Crick's 1958 hypothesis for how DNA codes for proteins.
Surprisingly, the 'Sanger' DNA sequencing method that we all know and (mostly) love wasn't actually developed until 1977!
###
Ryle AP et al. 1955. The disulphide bonds of insulin. Biochem. J. DOI: 10.1042/bj0600541