Omicly Weekly 8
January 21, 2024
Hey There!
Thanks for spending part of your Sunday with Omicly!
This week's headlines include:
1) Sequencing proteins with nanopores is hard. Engineered nanopores might make it a little easier.
2) The Proteome: It's a bit more complex than any of us would like
3) Did you know that there was a high quality diffraction of B-DNA made a year before Rosalind Franklin and Raymond Gosling generated photo 51?
Please enjoy!
Nanopores, what can't they do? Well, they're not very good at sequencing proteins. Yet.
‘Why would anyone want to sequence a protein?’
Because, they’re the functional molecules in our cells and the ultimate end product of our genome!
And when our genome gets damaged, it’s changes in the amount of protein, when those proteins are made, how they’re modified or the creation of mutated proteins that results in disease.
So, knowing what proteins are present and how they’re broken is important.
Luckily, sequencing proteins can tell us something about all of those things!
There are a couple ways to approach doing this, but one of the newest and most interesting methods for sequencing things is to use nanopores.
Currently, nanopores are usually made using proteins that create tiny holes in a membrane.
As molecules pass through these holes, the size and the shape of the molecule changes the amount of electrical current that can pass through the hole.
This change in current can be measured, and those signals can then be used to predict what was passing through the pore at any given time!
Oxford Nanopore have been very successful in refining this technique to sequence both DNA and RNA.
But, proteins offer a slightly new challenge.
To get DNA and RNA to pass through a pore, a positive charge is placed on one side of a nanopore.
This draws nucleic acids through the pore because they have a net negative charge.
But proteins are different.
They are composed of both positively and negatively charged amino acids, so pulling them through a pore with a positive or negative electrical field would just end up with them getting stuck!
Fortunately, smart people realized that there are other ways to pull (or push) molecules through a pore, one of them being electroosmotic flow (EOF).
That might sound complicated, but it really just means: use water as the carrier.
And that’s exactly what the author’s of today’s paper did!
They had to get pretty creative though, because these pore proteins aren't good at EOF without a little help.
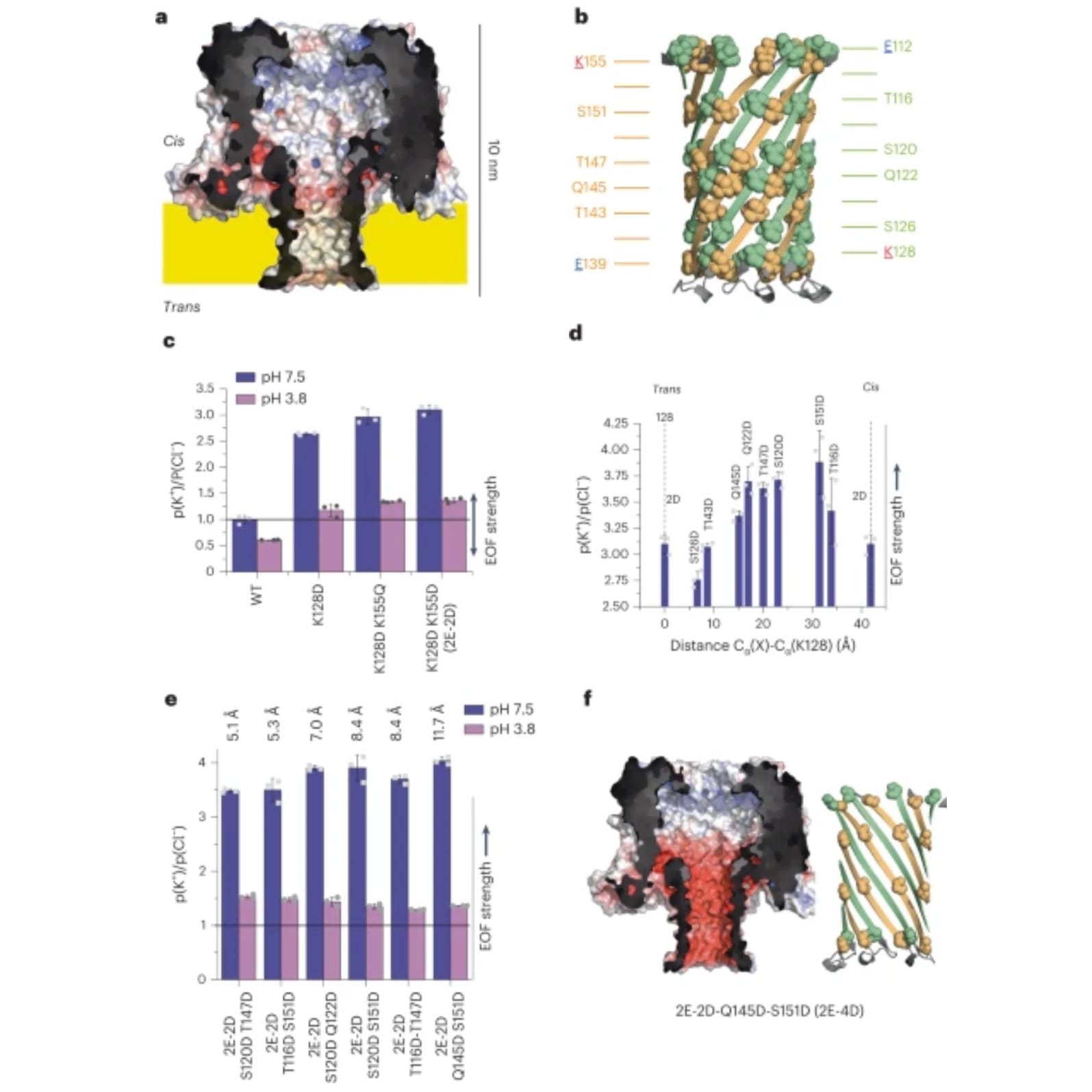
To do this, they specifically engineered a pore protein, CytK (a), and the barrel of the protein (b), to have negatively charged rings of amino acids to facilitate EOF when in the presence of an electric field.
The EOF performance of 2 (c), 3 (d), and 4 (e) rings was measured and it was found that 4 rings spaced 11.5 Å apart did the best.
The structure and charge of the winning pore, Q145D-S151D-CytK (2E-4D), can be seen in f.
Importantly, the authors went on to show that they can get proteins to pass completely through the pore (a fundamental requirement of any sequencer!) and generate current traces in the process.
But, converting that data into usable sequence is probably still many years out!
###
Sauciuc A, et al. 2023. Translocation of linearized full-length proteins through an engineered nanopore… Nature Biotech. DOI: 10.1038/s41587-023-01954-x
Translating the Proteome: There’s more to making proteins than just transcribing DNA into mRNA.

The biology 101 description of protein synthesis usually goes as follows:
RNA polymerase transcribes DNA into messenger RNA (mRNA).
mRNA is bound by a ribosome.
Bada bing bada boom.
Protein!
Except, there’s a lot of stuff that happens in-between the creation of mRNA and the translation of that message by a ribosome.
And much like DNA transcription, there are MULTIPLE levels of regulation that occur on the cellular level to make sure that the right proteins are made at the right time!
With that in mind, it's also important to note that mRNA abundance is not directly correlated with protein abundance for approximately 30% of genes.
So, just having mRNA around doesn’t necessarily mean that a protein is going to be made!
What the heck is going on here?
Translation Initiation: This is the first and arguably most regulated step! For an mRNA to be translated it has to be found and bound by a ribosome. Except ribosomes aren’t just bouncing around the cytoplasm (or endoplasmic reticulum) looking for naked mRNAs! Ribosomes are actually recruited by other other factors like the cap binding protein complex eIF4F (eukaryotic translation initiation factors) and the Poly-A Binding Protein (PABP) that binds the mRNA tail. These two protein complexes circularize the mRNA. That way, when a ribosome gets to the end, it’s back at the beginning to start all over again! However, this circularization process is highly regulated and even when a ribosome does find its way to one of these RNAs, there’s no guarantee that it will actually be translated! The ribosome itself is regulated, and for it to start the process of scanning for the translation initiation codon (the start site) it must be bound by a methionine loaded tRNA!
Translation Elongation: This is the phase of translation where all of the amino acids are added to the growing protein chain. However, how quickly those amino acids are added can be regulated and this can affect how much protein is made from any one message!
Location, Location, Location: Whether an mRNA is translated into protein depends a lot on where it is! This makes sense and is an important factor in the regulation of translation. If an RNA is no longer needed, it can be bound by factors that bring it to a processing body (P-body) for decay. These are little areas within the cytoplasm where RNAs are decapped and chopped up by nucleases. But if a cell doesn’t want to destroy an RNA, it can save it for later by moving it to a stress granule (like a p-body, just with less chopping!). This usually happens during stressful situations and translation can be completed when conditions improve.
microRNAs: These are small pieces of RNA that bind to the 3’-UTR (Untranslated Region) of an mRNA and mark it for death by the RNA-induced Silencing Complex (RISC). If an RNA is disappeared, it can’t be translated!
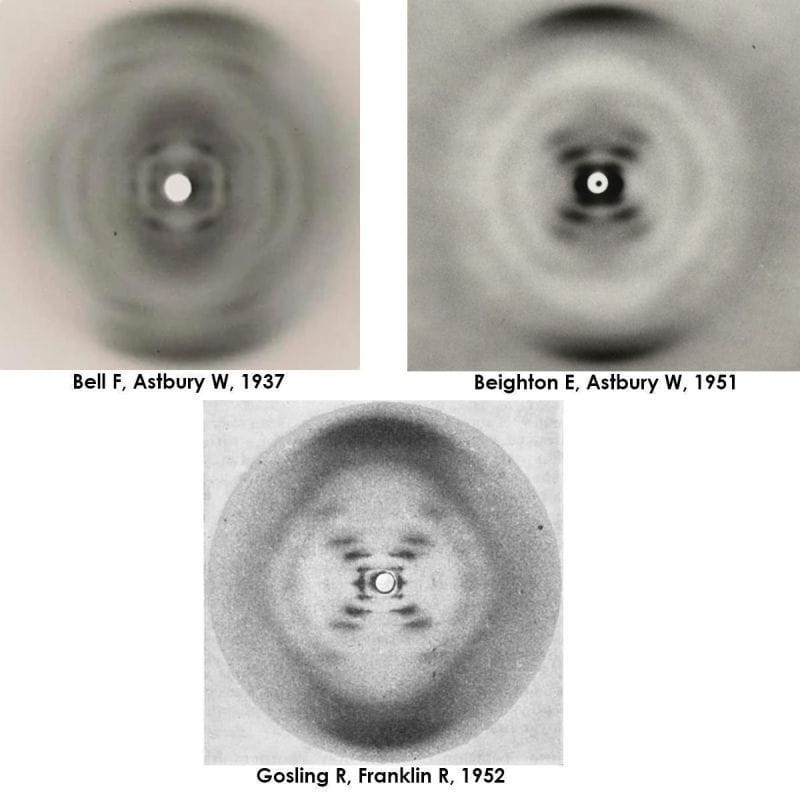
Elwyn Beighton and William Astbury generated a nearly flawless diffraction of B-DNA in 1951, a full year ahead of Franklin and Gosling. They never shared or published it.
As with everything on the path to the discovery of the DNA double-helix, timing was everything!
Well, timing, and knowing what you're looking at.
William Astbury might not be a name you've heard before but he's considered a founder of molecular biology.
He was one of the first to use x-ray diffraction to study protein structures.
He was a protege of William Bragg who in 1915 won the Nobel Prize, along with his son Lawrence, for the discovery that x-rays could be used to determine the location of atoms within a molecule.
What the Bragg's found was that if you blasted crystals with x-rays, the x-rays bounced off of the atoms in those molecules to create specific patterns on x-ray film.
Working backwards, they realized they could deduce from those patterns the structure of the underlying molecule!
However, by 1926, Bragg got bored with blasting simple molecules and tasked his graduate student, Astbury, with studying larger fibrous biological molecules like wool.
Since textiles were an important commodity, studying their properties and how to modify them to improve their commercial value was a big deal.
This led Astbury to start a lab in 1928 in textile physics where he made a name for himself diffracting just about every biological fiber in existence.
So, it should be no surprise that in 1937 he turned his attention to the most important biological fiber of all, DNA.
He, along with a talented graduate student, Florence Bell, created the first ever diffractions for DNA and they published the first proposed structure for DNA, what they referred to as a 'pile of pennies,' in 1938.
Unfortunately, Bell and Astbury's work on DNA was cut short by World War II and Astbury didn't return to the problem of DNA until the late 1940's.
But by 1951, Elwyn Beighton, a lab tech turned graduate student, had picked up where Bell left off and produced a nearly perfect diffraction of DNA.
Unbeknownst to anyone at the time, DNA could take on two forms: a totally dehydrated A form, a hydrated B form, or a mix of both depending on the humidity during drying.
The figure above is actually 3 figures. It has Bell's original diffraction of DNA (likely a mix of A and B), Beighton's significantly improved diffraction, and finally, photo 51, Franklin and Gosling's pristine diffraction of B-DNA.
All of the 'what ifs' aside, it's thought that Astbury was too preoccupied with protein structures to recognize the importance of Beighton's cruciform DNA diffraction.
The image was never published, and was lost to history.
Fortunately, the world only had to wait 2 additional years for the structure of DNA to be solved in 1953.
But, I wonder what other scientific breakthroughs have been filed away in dusty lab drawers?