Weekly Reading List: June 30,2025

Palm Beach Gardens Man wants to revive disgraced Theranos brand
A local inventor is trying to make a name for himself in a rather unusual way.

Eric Green was the first institute director forced out of NIH. He still hasn’t been told why
Eric
Green said the process leading to his ouster as a NIH institute
director by the Trump administration remains “shrouded in mystery”

Mapping the Transcriptional Landscape of Drug Responses in Primary Human Cells Using High-Throughput DRUG-seq
To advance our
understanding of drug action in physiologically-relevant systems, we
developed a high-throughput transcriptomic atlas of compound responses
in primary human cell types. Leveraging the scalable and cost-effective
Digital RNA with the pertUrbation of Genes (DRUG-seq) assay, we profiled
gene expression responses to 89 pharmacologically-active compounds
across six concentrations in four distinct primary cell types: aortic
smooth muscle cells (AoSMCs), skeletal muscle myoblasts (SkMMs), dermal
fibroblasts, and melanocytes.
Genetic disease risks of under-represented founder populations in New York City
Author
summary It is well recognized that genomic studies have been biased
towards individuals of European ancestry, and that obtaining medical
insights for populations under-represented in medical genomics is
crucial to achieve health equity. Here, we use genomic information to
identify networks of individuals in New York City who are distinctively
related to each other, allowing us to define populations with common
genetic ancestry based on genetic similarities rather than by
self-reported race or ethnicity. In our study of >25,000 New Yorkers,
we identified seven highly-interrelated founder populations, with 201
likely disease-causing variants with increased frequencies in specific
founder populations. Many of these population-specific variants are new
discoveries, despite their high frequency in founder populations.
Studying recent genetic ancestry can help reveal population-specific
disease insights that can help with early diagnosis, carrier screening,
and opportunities for targeted therapies that all help to reduce health
disparities in genomic medicine.

Predicting resistance to chemotherapy using chromosomal instability signatures - Nature Genetics
Here
the authors show that chromosomal instability signatures can predict
resistance to anthracycline-, taxane- and platinum-based
chemotherapeutics in breast, ovarian and prostate cancer and sarcoma.
Validation is performed through emulation of phase 2 and 3 clinical
trials using real-world data.

Paleolake geochronology supports Last Glacial Maximum (LGM) age for human tracks at White Sands, New Mexico
Discovery of human
footprints in alluvium dated to the Last Glacial Maximum (LGM) at White
Sands, New Mexico, was a notable step in understanding the initial
peopling of the Americas, but that work was met with criticism focused
on the reliability of the materials used in the radiocarbon dating
(seeds of Ruppia and pollen). This paper reports on an independent study
of the chronology of a previously unrecognized stratigraphic record of
paleolake Otero that is directly traceable into the track-bearing
alluvium.
Millions of children at risk as vaccination uptake stalls
A
global study finds large numbers of children are unvaccinated against
diseases like measles, tuberculosis and polio, which makes outbreaks
more likely.
llumina to Acquire SomaLogic Assets from Standard BioTools for up to $425M
Illumina said Monday
that it will acquire proteomics technology originally developed by
SomaLogic, among other assets, from Standard BioTools for $350 million
in cash. The deal also includes up to $75 million in near-term milestone
cash payments and performance-based royalties.
RFK Jr. defends proposed HHS budget as Democrats slam cuts, gutting of CDC vaccine panel
Department
of Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. |
HHS Secretary Robert F. Kennedy Jr. defended his reorganization of the
agency and the proposed fiscal 2026 budget that cuts funding by 25%
during a hearing Tuesday of the House Energy and Commerce Health
subcommittee.

Cassidy calls to delay meeting of CDC’s vaccine panel in challenge to RFK Jr.
A key GOP senator is calling for the CDC’s vaccine meeting to be postponed after RFK Jr. shook up the panel.

GRK-biased adrenergic agonists for the treatment of type 2 diabetes and obesity
Biased agonism of G
protein-coupled receptors (GPCRs) offers potential for safer
medications. Current efforts have explored the balance between G
proteins and β-arrestin; however, other transducers like GPCR kinases
(GRKs) remain understudied. GRK2 is essential for β2 adrenergic receptor
(β2AR)-mediated glucose uptake, but β2AR agonists are considered poor
clinical candidates for glycemic management due to Gs/cyclic AMP
(cAMP)-induced cardiac side effects and β-arrestin-dependent
desensitization. Using ligand-based virtual screening and chemical
evolution, we developed pathway-selective agonists of β2AR that prefer
GRK coupling.
Infectome
analysis of bat kidneys from Yunnan province, China, reveals novel
henipaviruses related to Hendra and Nipah viruses and prevalent
bacterial and eukaryotic microbes
Author
summary Although extensive investigations have been conducted on the
bat virome, most studies have focused on fecal samples, leaving other
tissues, such as the kidney, largely unexplored. However, the kidney can
harbor important zoonotic pathogens, including the highly pathogenic
Hendra and Nipah viruses, and genomic evidence of henipaviruses in bats
from China has remained undocumented. In this study, we report the first
detection of two novel henipavirus genomes from bat kidneys in China,
one of which is the closest known relative of pathogenic henipaviruses
identified to date. Beyond virome analysis, our study also examined
highly prevalent bacteria and eukaryotic microbes, identifying those
potentially relevant to bat infections. Overall, these findings provide
valuable insights into the infectome of the bat kidney, highlighting the
need for broader microbial surveillance beyond the gastrointestinal
tract.

Clinical report outlines how, why to pursue genetic diagnosis for GDD/ID Free
Global developmental
delay and intellectual disability (GDD/ID) in children are common
concerns primary care pediatricians face. GDD/ID have diverse
etiologies, but genetic disorders account for a substantial percentage.
Establishing a genetic diagnosis provides multiple benefits for the
patient and family, including improvements in patient care and
determination of recurrence risk.
AlphaGenome: AI for better understanding the genome
Introducing
a new, unifying DNA sequence model that advances regulatory
variant-effect prediction and promises to shed new light on genome
function — now available via API.

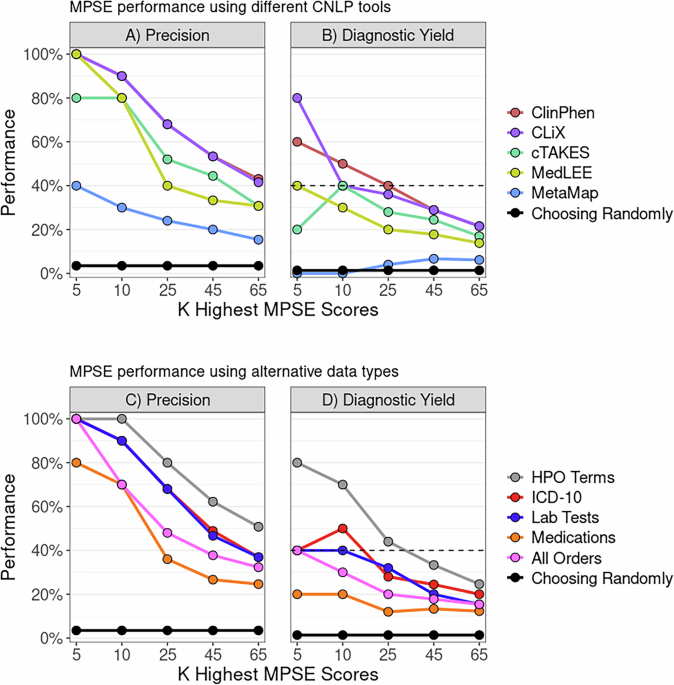
MPSE identifies newborns for whole genome sequencing within 48 h of NICU admission - npj Genomic Medicine
npj Genomic Medicine - MPSE identifies newborns for whole genome sequencing within 48 h of NICU admission
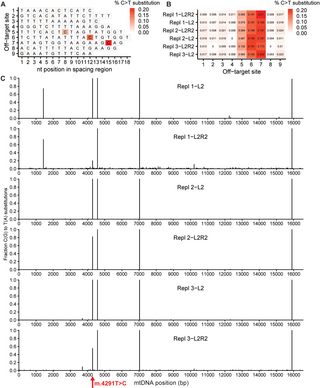
Correction of pathogenic mitochondrial DNA in patient-derived disease models using mitochondrial base editors
Recent
developments in base editing technologies enable the correction of
mutations in the mitochondrial genome, but its therapeutic potential
remains unclear. This proof-of-principle study shows that mitochondrial
base editing can functionally create and correct mitochondrial
pathogenic mutations in patient-derived cells.
CDC panel, newly remade by RFK Jr., questions vaccine evidence
New
members of the ACIP panel raised questions about the evidence
supporting COVID vaccines, and signaled plans to look at other
established shots, like those for measles and hepatitis B.

Judge overturns NSF’s 15% cap on reimbursing indirect costs of research
Agency’s action deemed in violation of federal law

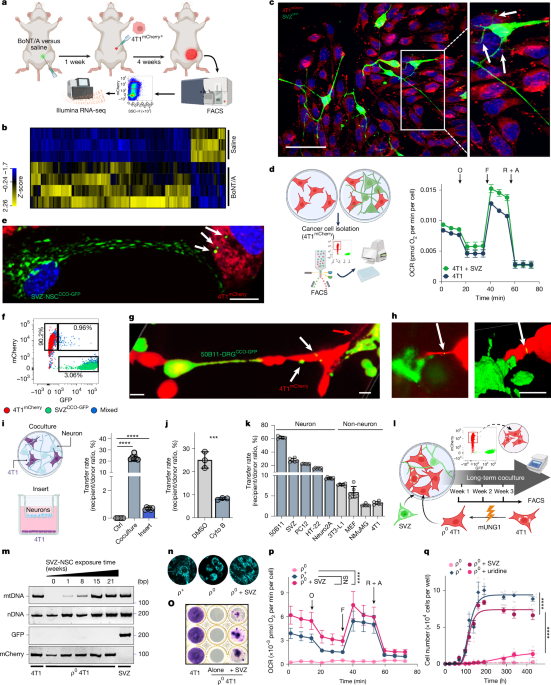
Nerve-to-cancer transfer of mitochondria during cancer metastasis - Nature
A
study reports the development of a method to trace intercellular
transfer of mitochondria, and demonstrates that cancer cells that
receive mitochondria from neurons have enhanced metastatic capabilities.
uv, part 4: uv with Jupyter
Using uv with Jupyter: demo using polars and seaborn for analysis and visualization.
Artificial Intelligence in Proteomics Workflows Takes off
Artificial intelligence
and deep learning are carving out substantial roles in proteomic
workflows, improving peptide and protein identifications, advancing
applications like de novo sequencing, and allowing scientists to more
accurately and comprehensively model biological systems.
French scientists discover new blood type in Guadeloupe woman
Thanks to DNA sequencing, the discovery of new blood groups has accelerated in recent years.

ACMG SF v3.3 list for reporting of secondary findings in clinical exome and genome sequencing
The American College of
Medical Genetics and Genomics (ACMG) previously published guidance for
reporting secondary findings (SFs) in the context of clinical exome and
genome sequencing.1-7 The ACMG Secondary Findings Working Group (SFWG)
and Board of Directors (BODs) have agreed that the list of recommended
genes should be updated annually and with an ongoing goal of maintaining
this as a minimum list. Reporting of SFs should be considered neither a
replacement for indication-based diagnostic clinical genetic testing
nor a form of population screening.
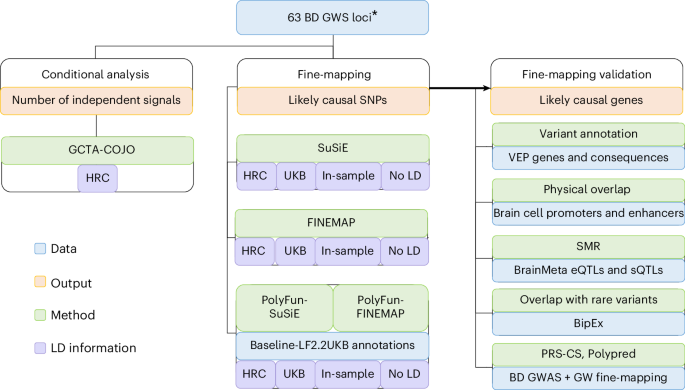
Fine-mapping genomic loci refines bipolar disorder risk genes - Nature Neuroscience
This
study used fine-mapping to analyze genetic regions associated with
bipolar disorder, identifying specific risk genes and providing new
insights into the biology of the condition that may guide future
research and treatment approaches.
The New ‘Razor Blade Throat’ Nimbus COVID Variant: Symptoms, Incubation Period and When to Test | KQED
A
new COVID variant called “Nimbus” is spreading. Here’s what you need to
know about the incubation period, symptoms (including “razor blade
throat”) and when to take a test.

Supreme Court Preserves ACA Preventive Requirements for Private Plans; Rejects Challenge to USPSTF
The
Affordable Care Act of 2010 requires private insurance to cover
preventive benefits endorsed by the USPTF. The legitimacy of the USPS…

Common and rare genetic variants show network convergence for a majority of human traits
While
both common and rare variants contribute to the genetic etiology of
complex traits, whether their impacts manifest through the same effector
genes and molecular mechanisms is not well understood. Here, we
systematically analyze common and rare variants associated with each of
373 phenotypic traits within a large biological knowledge network of
gene and protein interactions. While common and rare variants implicate
few shared genes, they converge on shared molecular networks for more
than 75% of traits. We demonstrate that the strength of this convergence
is influenced by core factors such as trait heritability, gene
mutational constraints, and tissue specificity. Using neuropsychiatric
traits as examples, we show that common and rare variants impact shared
functions across multiple levels of biological organization. These
findings underscore the importance of integrating variants across the
frequency spectrum and establish a foundation for network-based
investigations of the genetics of diverse human diseases and phenotypes.
### Competing Interest Statement T.I. is a co-founder, member of the
advisory board, and has an equity interest in Data4Cure and Serinus
Biosciences. T.I. is a consultant for and has an equity interest in
Ideaya Biosciences and Eikon Therapeutics. The terms of these
arrangements have been reviewed and approved by the University of
California, San Diego, in accordance with its conflict-of-interest
policies. ### Funding Statement This work was supported by the following
grants from the National Institutes of Health: NIMH U01 MH115747 to
T.I., and NIDA P50 DA037844 to T.I. This work was also supported by the
following grants from the California Institute for Regenerative
Medicine: ReMIND DISC4-16322 and ReMIND DISC4-16377. ### Author
Declarations I confirm all relevant ethical guidelines have been
followed, and any necessary IRB and/or ethics committee approvals have
been obtained. Yes The details of the IRB/oversight body that provided
approval or exemption for the research described are given below: All
human data utilized in this study were openly available before the
initiation of the study. Summary results from previously published
genome wide association studies (GWAS) were sourced from the GWAS
Catalog (https://www.ebi.ac.uk/gwas/api/search/downloads/alternative).
Accession numbers for the studies used can be found in Supplemental
Table 1. Summary results from previously published gene-level rare
variant studies were sourced from the Rare Variant Association
Repository (RAVAR,
http://www.ravar.bio/api/download/static/gene_fulltable.txt).
Publication identifiers (PMIDs) for the studies used can be found in
Supplemental Table 1. I confirm that all necessary patient/participant
consent has been obtained and the appropriate institutional forms have
been archived, and that any patient/participant/sample identifiers
included were not known to anyone (e.g., hospital staff, patients or
participants themselves) outside the research group so cannot be used to
identify individuals. Yes I understand that all clinical trials and any
other prospective interventional studies must be registered with an
ICMJE-approved registry, such as ClinicalTrials.gov. I confirm that any
such study reported in the manuscript has been registered and the trial
registration ID is provided (note: if posting a prospective study
registered retrospectively, please provide a statement in the trial ID
field explaining why the study was not registered in advance). Yes I
have followed all appropriate research reporting guidelines, such as any
relevant EQUATOR Network research reporting checklist(s) and other
pertinent material, if applicable. Yes All data produced in the present
work are contained in the manuscript
<https://www.ebi.ac.uk/gwas/api/search/downloads/alternative>
<http://www.ravar.bio/api/download/static/gene_fulltable.txt>